

Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay
- PMID: 15687290
- PMCID: PMC546528
- DOI: 10.1101/gr.3185605
Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay
Abstract
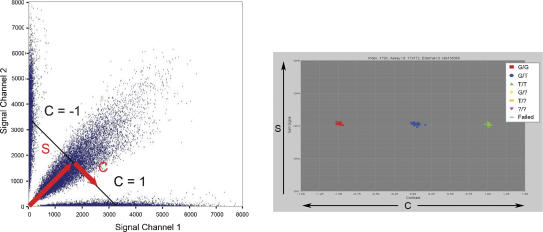
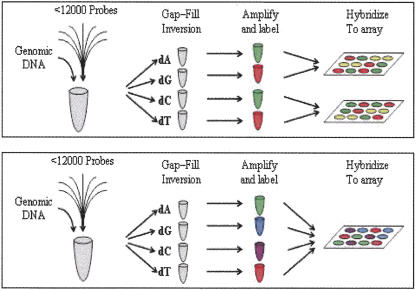
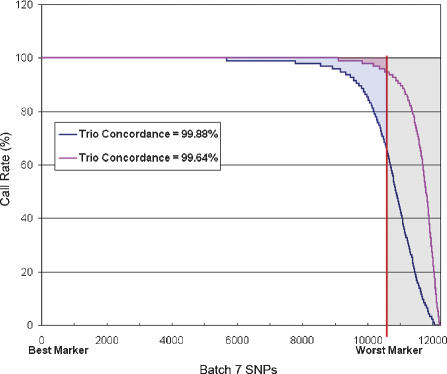
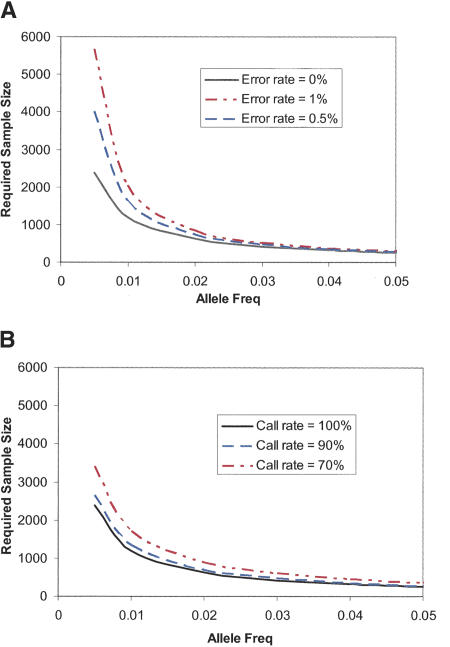
Large-scale genetic studies are highly dependent on efficient and scalable multiplex SNP assays. In this study, we report the development of Molecular Inversion Probe technology with four-color, single array detection, applied to large-scale genotyping of up to 12,000 SNPs per reaction. While generating 38,429 SNP assays using this technology in a population of 30 trios from the Centre d'Etude Polymorphisme Humain family panel as part of the International HapMap project, we established SNP conversion rates of approximately 90% with concordance rates >99.6% and completeness levels >98% for assays multiplexed up to 12,000plex levels. Furthermore, these individual metrics can be "traded off" and, by sacrificing a small fraction of the conversion rate, the accuracy can be increased to very high levels. No loss of performance is seen when scaling from 6,000plex to 12,000plex assays, strongly validating the ability of the technology to suppress cross-reactivity at high multiplex levels. The results of this study demonstrate the suitability of this technology for comprehensive association studies that use targeted SNPs in indirect linkage disequilibrium studies or that directly screen for causative mutations.
Figures
References
-
- Altshuler, D., Hirschhorn, J.N., Klannemark, M., Lindgren, C.M., Vohl, M.C., Nemesh, J., Lane, C.R., Schaffner, S.F., Bolk, S., Brewer, C., et al. 2000. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 26: 76-80. - PubMed
-
- Grossman, P.D., Bloch, W., Brinson, E., Chang, C.C., Eggerding, F.A., Fung, S., Iovannisci, D.M., Woo, S., Winn-Deen, E.S., and Iovannisci, D.A. 1994. High-density multiplex detection of nucleic acid sequences: Oligonucleotide ligation assay and sequence-coded separation. Nucleic Acids Res. 22: 4527-4534. - PMC - PubMed
-
- Hardenbol, P., Baner, J., Jain, M., Nilsson, M., Namsaraev, E.A., Karlin-Neumann, G.A., Fakhrai-Rad, H., Ronaghi, M., Willis, T.D., Landegren, U., et al. 2003. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 21: 673-678. - PubMed
WEB SITE REFERENCES
-
- www.hapmap.org; Chromosome 12 data, HapMap Project Web site.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources