

Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury
- PMID: 15693986
- PMCID: PMC1065095
- DOI: 10.1186/cc2950
Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury
Abstract
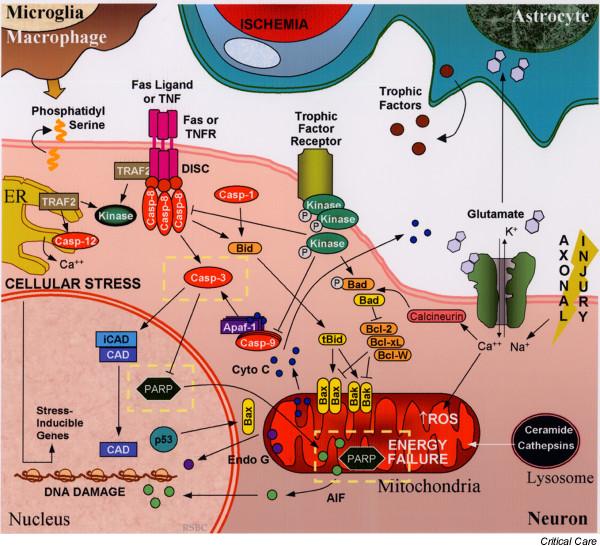
Apoptosis, or programmed cell death, is a physiological form of cell death that is important for normal embryologic development and cell turnover in adult organisms. Cumulative evidence suggests that apoptosis can also be triggered in tissues without a high rate of cell turnover, including those within the central nervous system (CNS). In fact, a crucial role for apoptosis in delayed neuronal loss after both acute and chronic CNS injury is emerging. In the current review we summarize the growing evidence that apoptosis occurs after traumatic brain injury (TBI), from experimental models to humans. This includes the identification of apoptosis after TBI, initiators of apoptosis, key modulators of apoptosis such as the Bcl-2 family, key executioners of apoptosis such as the caspase family, final pathways of apoptosis, and potential therapeutic interventions for blocking neuronal apoptosis after TBI.
Figures
References
-
- Jager TE, Weiss HB, Coben JH, Pepe PE. Traumatic brain injuries evaluated in U.S. emergency departments, 1992–1994. Acad Emerg Med. 2000;7:134–140. - PubMed
-
- Adekoya N, Thurman DJ, White DD, Webb KW. Surveillance for traumatic brain injury deaths – United States, 1989–1998. Morb Mort Weekly Rept Surveill Summ. 2002;51:1–14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
