

Homology-extended sequence alignment
- PMID: 15699183
- PMCID: PMC549400
- DOI: 10.1093/nar/gki233
Homology-extended sequence alignment
Abstract
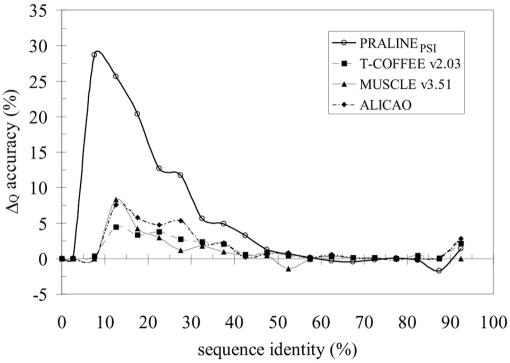
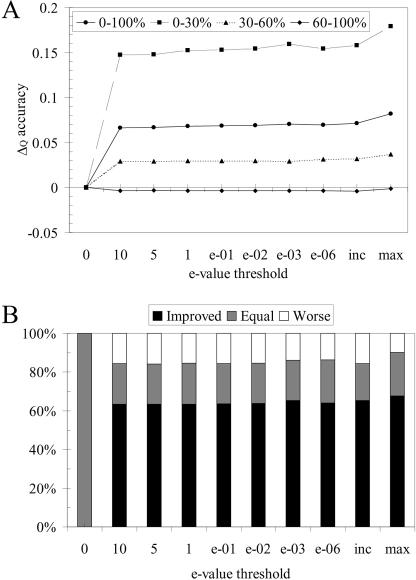
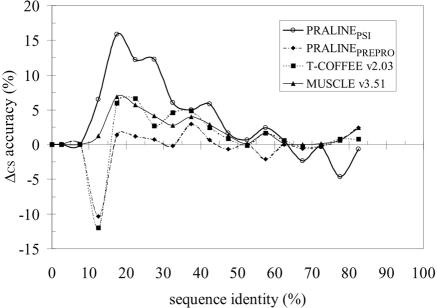
We present a profile-profile multiple alignment strategy that uses database searching to collect homologues for each sequence in a given set, in order to enrich their available evolutionary information for the alignment. For each of the alignment sequences, the putative homologous sequences that score above a pre-defined threshold are incorporated into a position-specific pre-alignment profile. The enriched position-specific profile is used for standard progressive alignment, thereby more accurately describing the characteristic features of the given sequence set. We show that owing to the incorporation of the pre-alignment information into a standard progressive multiple alignment routine, the alignment quality between distant sequences increases significantly and outperforms state-of-the-art methods, such as T-COFFEE and MUSCLE. We also show that although entirely sequence-based, our novel strategy is better at aligning distant sequences when compared with a recent contact-based alignment method. Therefore, our pre-alignment profile strategy should be advantageous for applications that rely on high alignment accuracy such as local structure prediction, comparative modelling and threading.
Figures


References
-
- Simossis V.A., Kleinjung J., Heringa J. In: Current Protocols in Bioinformatics. Baxevanis A.D., editor. NY: John Wiley; 2003. pp. 3.7.1–3.7.25. - PubMed
-
- Needleman S.B., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. - PubMed
-
- Feng D.F., Doolittle R.F. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 1987;25:351–360. - PubMed
-
- Barker W.C., Ketcham L.K., Dayhoff M.O. A comprehensive examination of protein sequences for evidence of internal gene duplication. J. Mol. Evol. 1978;10:265–281. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
