

Impaired vasodilation by red blood cells in sickle cell disease
- PMID: 15699345
- PMCID: PMC548996
- DOI: 10.1073/pnas.0409876102
Impaired vasodilation by red blood cells in sickle cell disease
Abstract
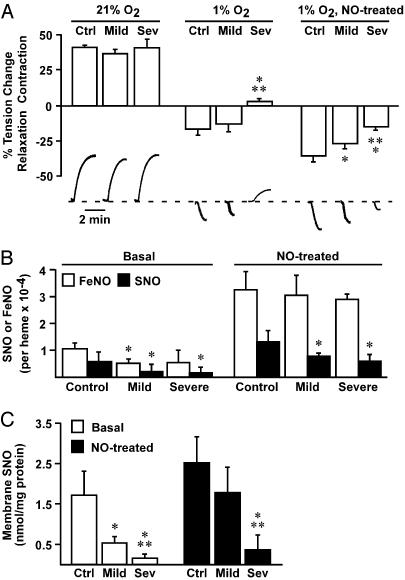
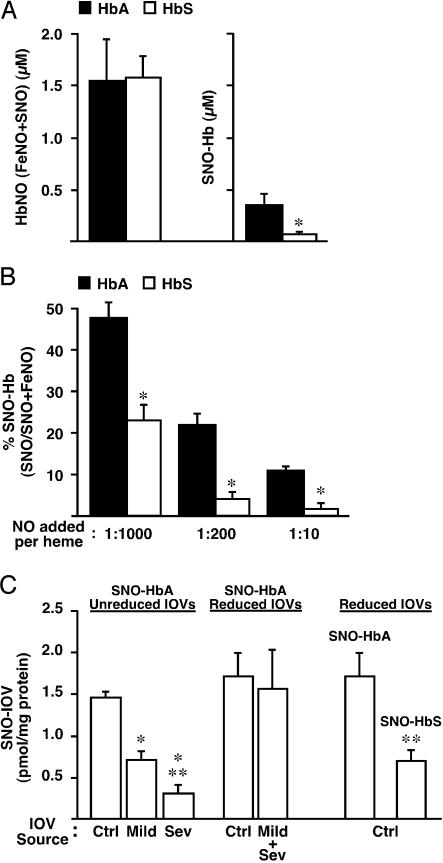
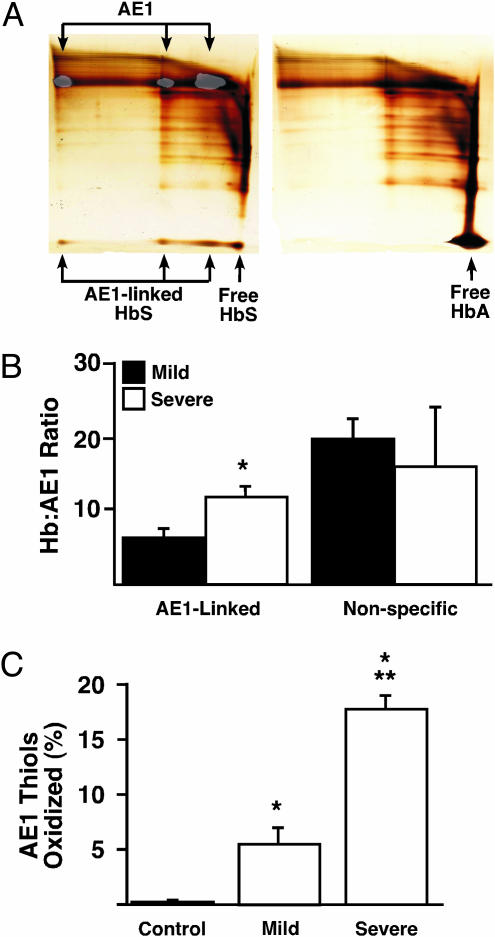
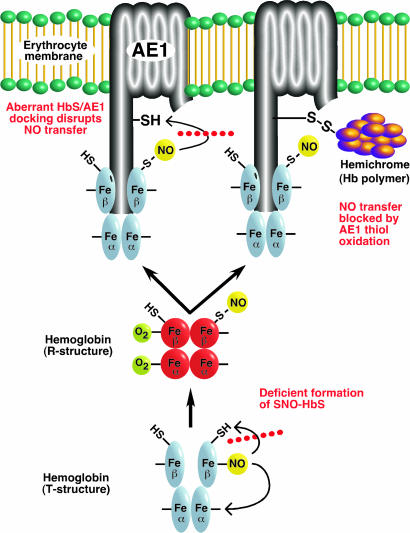
Red blood cells (RBCs) have been ascribed a unique role in dilating blood vessels, which requires O2-regulated binding and bioactivation of NO by Hb and transfer of NO equivalents to the RBC membrane. Vasoocclusion in hypoxic tissues is the hallmark of sickle cell anemia. Here we show that sickle cell Hb variant S (HbS) is deficient both in the intramolecular transfer of NO from heme iron (iron nitrosyl, FeNO) to cysteine thiol (S-nitrosothiol, SNO) that subserves bioactivation, and in transfer of the NO moiety from S-nitrosohemoglobin (SNO-HbS) to the RBC membrane. As a result, sickle RBCs are deficient in membrane SNO and impaired in their ability to mediate hypoxic vasodilation. Further, the magnitudes of these impairments correlate with the clinical severity of disease. Thus, our results suggest that abnormal RBC vasoactivity contributes to the vasoocclusive pathophysiology of sickle cell anemia, and that the phenotypic variation in expression of the sickle genotype may be explained, in part, by variable deficiency in RBC processing of NO. More generally, our findings raise the idea that defective NO processing may characterize a new class of hemoglobinopathy.
Figures
References
-
- McMahon, T. J., Moon, R. E., Luchsinger, B. P., Carraway, M. S., Stone, A. E., Stolp, B. W., Gow, A. J., Pawloski, J. R., Watke, P., Singel, D. J., et al. (2002) Nat. Med. 8, 711-717. - PubMed
-
- Pawloski, J. R., Swaminathan, R. V. & Stamler, J. S. (1998) Circulation 97, 263-267. - PubMed
-
- Pawloski, J. R., Hess, D. T. & Stamler, J. S. (2001) Nature 409, 622-626. - PubMed
-
- James, P. E., Lang, D., Tufnell-Barrett, T., Milsom, A. B. & Frenneaux, M. P. (2004) Circ. Res. 94, 976-983. - PubMed
-
- Datta, B., Tufnell-Barrett, T., Bleasdale, R. A., Jones, C. J., Beeton, I., Paul, V., Frenneaux, M. & James, P. (2004) Circulation 109, 1339-1342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
