

Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons
- PMID: 15703402
- PMCID: PMC6725987
- DOI: 10.1523/JNEUROSCI.3523-04.2005
Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons
Abstract
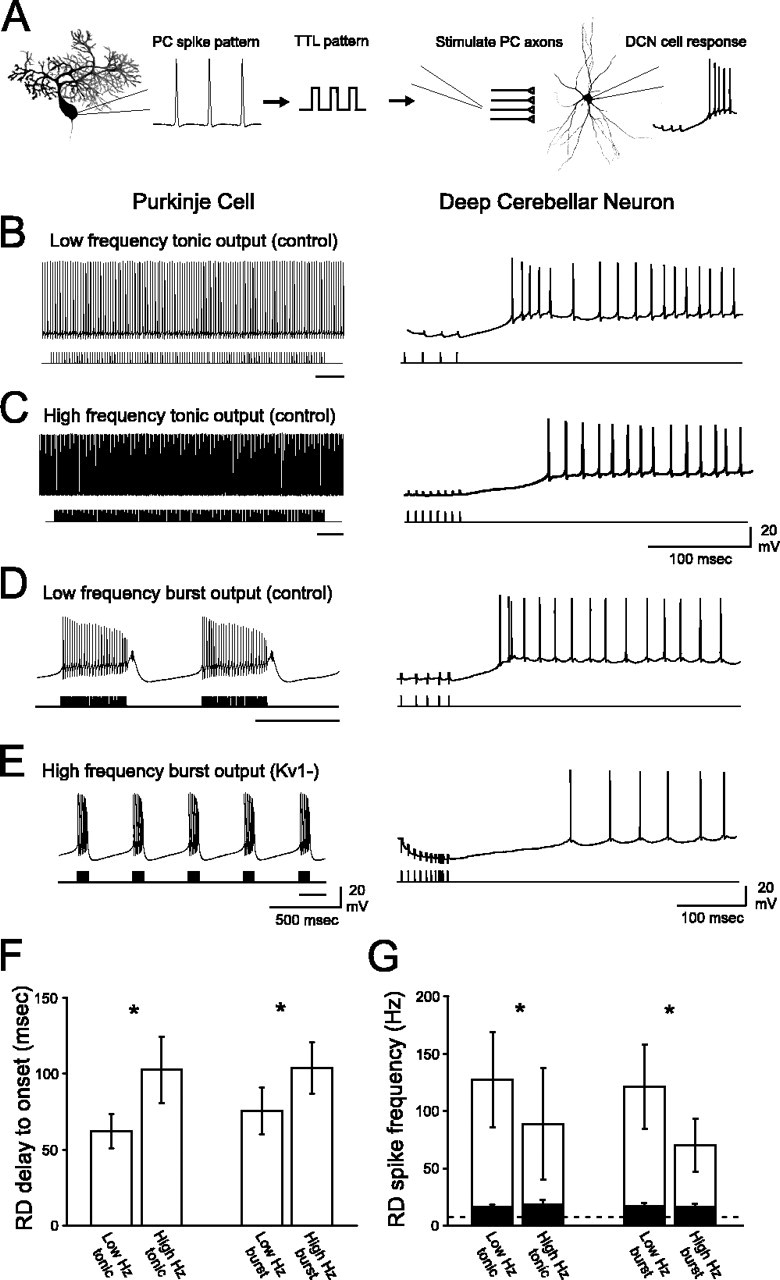
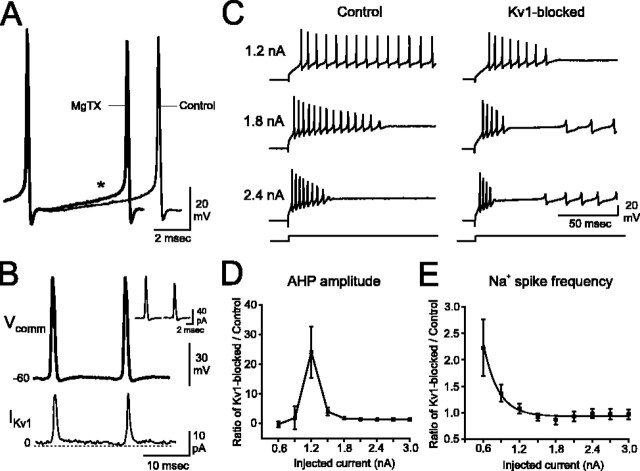
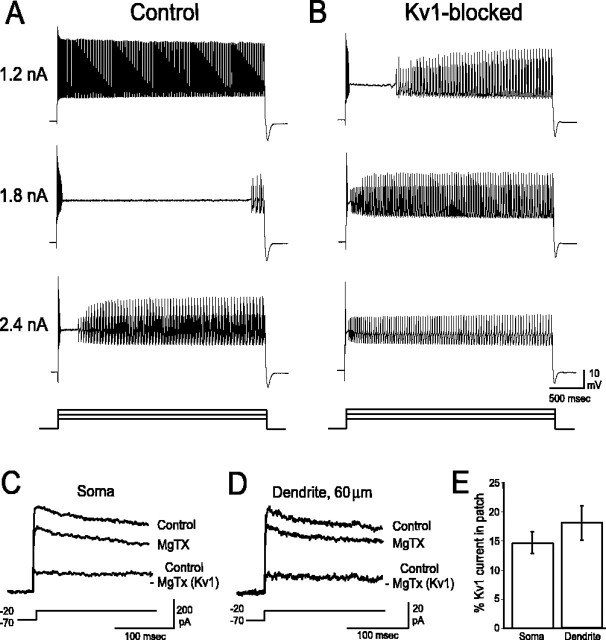
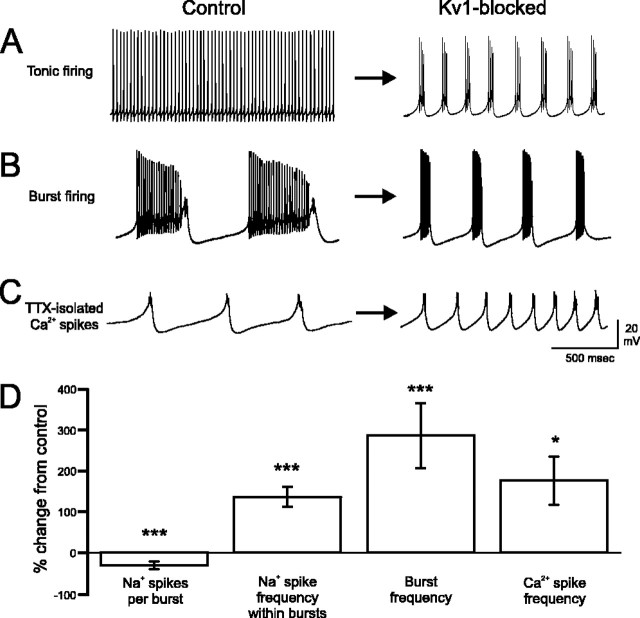
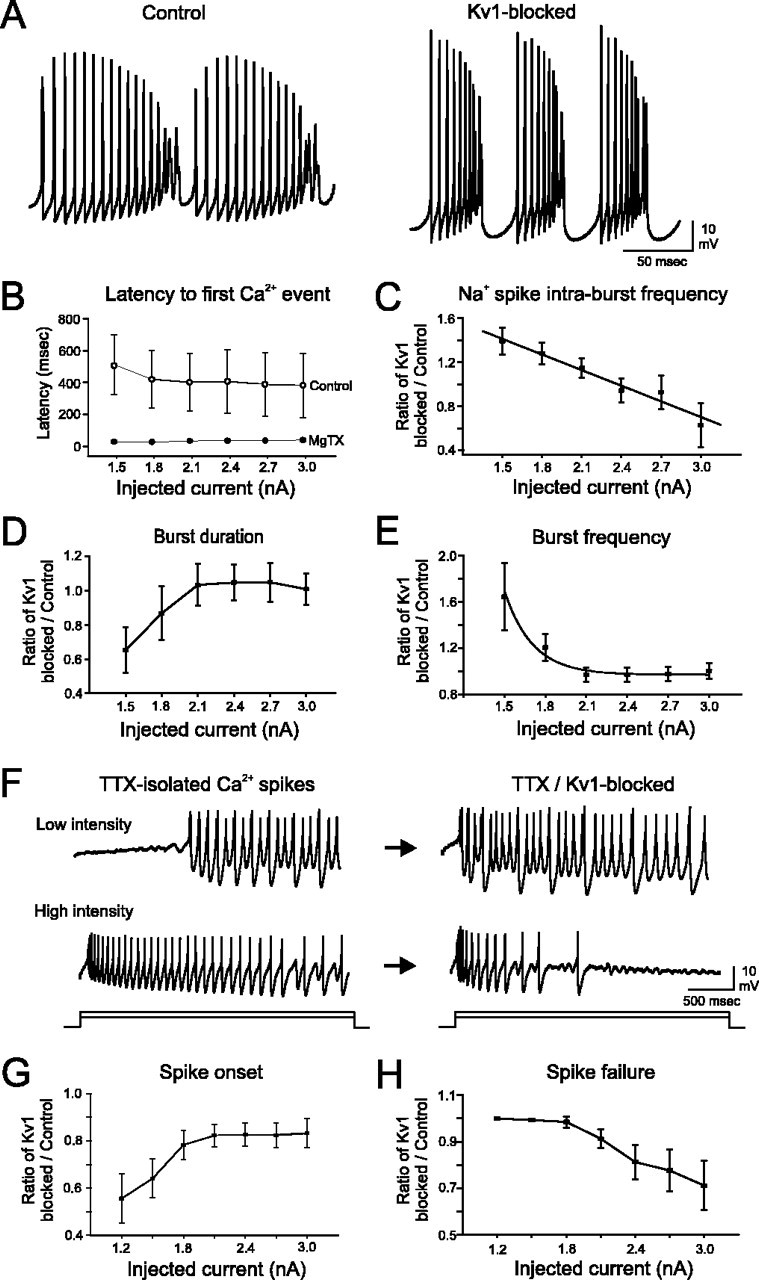
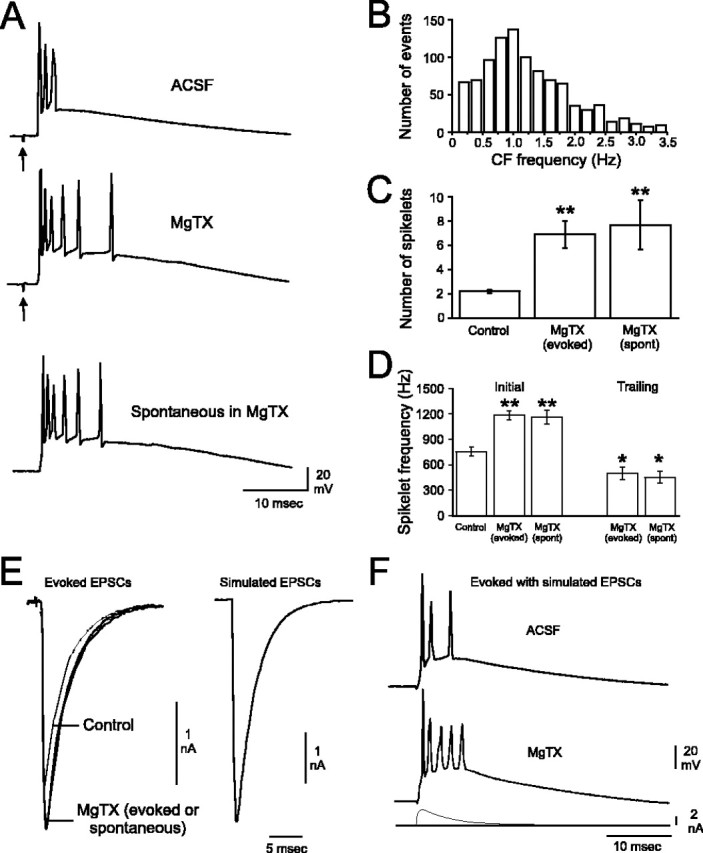
Purkinje cells (PCs) generate the sole output of the cerebellar cortex and govern the timing of action potential discharge from neurons of the deep cerebellar nuclei (DCN). Here, we examine how voltage-gated Kv1 K+ channels shape intrinsically generated and synaptically controlled behaviors of PCs and address how the timing of DCN neuron output is modulated by manipulating PC Kv1 channels. Kv1 channels were studied in cerebellar slices at physiological temperatures with Kv1-specific toxins. Outside-out voltage-clamp recordings indicated that Kv1 channels are present in both somatic and dendritic membranes and are activated by Na+ spike-clamp commands. Whole-cell current-clamp recordings revealed that Kv1 K+ channels maintain low frequencies of Na+ spike and Ca-Na burst output, regulate the duration of plateau potentials, and set the threshold for Ca2+ spike discharge. Kv1 channels shaped the characteristics of climbing fiber (CF) responses evoked by extracellular stimulation or intracellular simulated EPSCs. In the presence of Kv1 toxins, CFs discharged spontaneously at approximately 1 Hz. Finally, "Kv1-intact" and "Kv1-deficient" PC tonic and burst outputs were converted to stimulus protocols and used as patterns to stimulate PC axons and synaptically activate DCN neurons. We found that the Kv1-intact patterns facilitated short-latency and high-frequency DCN neuron rebound discharges, whereas DCN neuron output timing was markedly disrupted by the Kv1-deficient stimulus protocols. Our results suggest that Kv1 K+ channels are critical for regulating the excitability of PCs and CFs and optimize the timing of PC outputs to generate appropriate discharge patterns in postsynaptic DCN neurons.
Figures
Similar articles
-
Kv3 K+ channels enable burst output in rat cerebellar Purkinje cells.Eur J Neurosci. 2004 Aug;20(3):729-39. doi: 10.1111/j.1460-9568.2004.03539.x. Eur J Neurosci. 2004. PMID: 15255983
-
Iberiotoxin-sensitive large conductance Ca2+ -dependent K+ (BK) channels regulate the spike configuration in the burst firing of cerebellar Purkinje neurons.Brain Res. 2008 May 30;1212:1-8. doi: 10.1016/j.brainres.2008.03.030. Epub 2008 Mar 27. Brain Res. 2008. PMID: 18439989
-
Physiological role of dendrotoxin-sensitive K+ channels in the rat cerebellar Purkinje neurons.Physiol Res. 2007;56(6):807-813. doi: 10.33549/physiolres.931041. Epub 2006 Nov 6. Physiol Res. 2007. PMID: 17087603
-
Activity-dependent maturation of climbing fiber to Purkinje cell synapses during postnatal cerebellar development.Cerebellum. 2012 Jun;11(2):449-50. doi: 10.1007/s12311-011-0337-3. Cerebellum. 2012. PMID: 22194041 Review.
-
Rebound discharge in deep cerebellar nuclear neurons in vitro.Cerebellum. 2010 Sep;9(3):352-74. doi: 10.1007/s12311-010-0168-7. Cerebellum. 2010. PMID: 20396983 Free PMC article. Review.
Cited by
-
Interaction between Purkinje cells and inhibitory interneurons may create adjustable output waveforms to generate timed cerebellar output.PLoS One. 2008 Jul 23;3(7):e2770. doi: 10.1371/journal.pone.0002770. PLoS One. 2008. PMID: 18648667 Free PMC article.
-
The Roles of the Olivocerebellar Pathway in Motor Learning and Motor Control. A Consensus Paper.Cerebellum. 2017 Feb;16(1):230-252. doi: 10.1007/s12311-016-0787-8. Cerebellum. 2017. PMID: 27193702 Free PMC article.
-
Posthearing developmental refinement of temporal processing in principal neurons of the medial superior olive.J Neurosci. 2005 Aug 31;25(35):7887-95. doi: 10.1523/JNEUROSCI.1016-05.2005. J Neurosci. 2005. PMID: 16135745 Free PMC article.
-
Dendritic spikes mediate negative synaptic gain control in cerebellar Purkinje cells.Proc Natl Acad Sci U S A. 2010 Dec 21;107(51):22284-9. doi: 10.1073/pnas.1008605107. Epub 2010 Dec 3. Proc Natl Acad Sci U S A. 2010. PMID: 21131572 Free PMC article.
-
Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling.Trends Neurosci. 2010 May;33(5):211-9. doi: 10.1016/j.tins.2010.02.005. Epub 2010 Mar 11. Trends Neurosci. 2010. PMID: 20226542 Free PMC article.
References
-
- Aizenman CD, Linden DJ (1999) Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol 82: 1697-1709. - PubMed
-
- Aizenman CD, Manis PB, Linden DJ (1998) Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron 21: 827-835. - PubMed
-
- Albus JS (1971) A theory of cerebellar function. Math Biosci 10: 25-61.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous