

Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance
- PMID: 15705718
- PMCID: PMC552942
- DOI: 10.1073/pnas.0409770102
Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance
Abstract
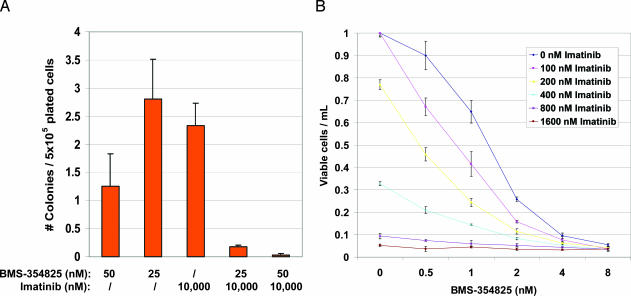
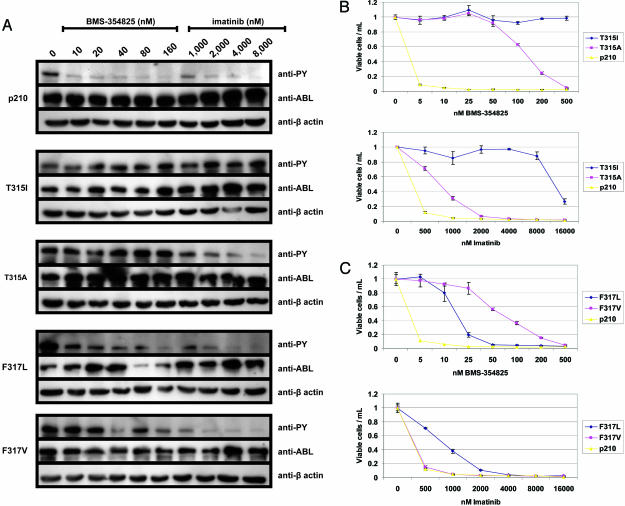
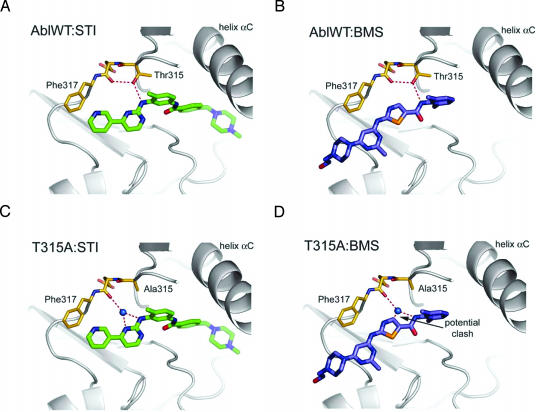
Structural studies suggest that most point mutations in the BCR-ABL kinase domain cause resistance to the ABL kinase inhibitor imatinib by impairing the flexibility of the kinase domain, restricting its ability to adopt the inactive conformation required for optimal imatinib binding, rather than by directly interfering with drug contact residues. BMS-354825, currently in clinical development for imatinib-resistant chronic myelogenous leukemia, is a dual SRC/ABL kinase inhibitor that binds ABL in both the active and inactive conformation. To examine the potential role of conformational binding properties in drug resistance, we mapped the mutations in BCR-ABL capable of conferring resistance to BMS-354825. Through saturation mutagenesis, we identified 10 such BCR-ABL mutations, 8 of which occurred at drug contact residues. Some mutants were unique to BMS-354825, whereas others also conferred imatinib resistance. Remarkably, the identity of the amino acid substitution at either of two contact residues differentially affects sensitivity to imatinib or BMS-354825. The combination of imatinib plus BMS-354825 greatly reduced the recovery of drug-resistant clones. Our findings provide further rationale for considering kinase conformation in the design of kinase inhibitors against cancer targets.
Figures
References
-
- Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B. & Kuriyan, J. (2000) Science 289, 1938-1942. - PubMed
-
- Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., Lydon, N. B., Kantarjian, H., Capdeville, R., Ohno-Jones, S. & Sawyers, C. L. (2001) N. Engl. J. Med. 344, 1031-1037. - PubMed
-
- O'Brien, S. G., Guilhot, F., Larson, R. A., Gathmann, I., Baccarani, M., Cervantes, F., Cornelissen, J. J., Fischer, T., Hochhaus, A., Hughes, T., et al. (2003) N. Engl. J. Med. 348, 994-1004. - PubMed
-
- Donato, N. J., Wu, J. Y., Stapley, J., Lin, H., Arlinghaus, R., Aggarwal, B. B., Shishodia, S., Albitar, M., Hayes, K., Kantarjian, H., et al. (2004) Cancer Res. 64, 672-677. - PubMed
-
- Donato, N. J., Wu, J. Y., Stapley, J., Gallick, G., Lin, H., Arlinghaus, R. & Talpaz, M. (2003) Blood 101, 690-698. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
