

Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer's disease
- PMID: 15705720
- PMCID: PMC549455
- DOI: 10.1073/pnas.0408500102
Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer's disease
Abstract
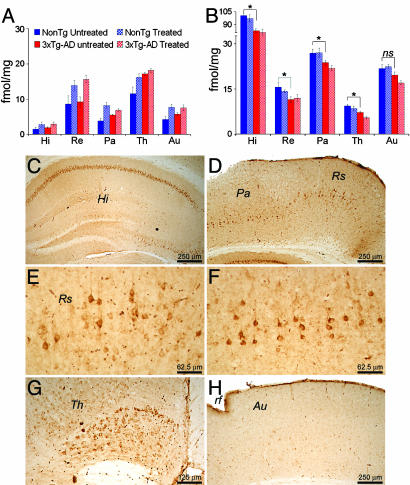
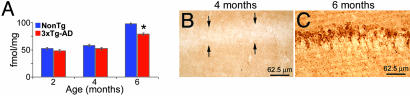
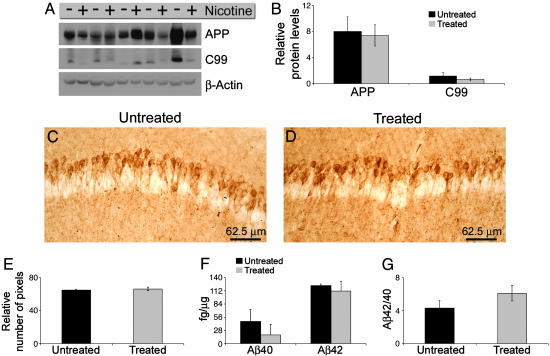
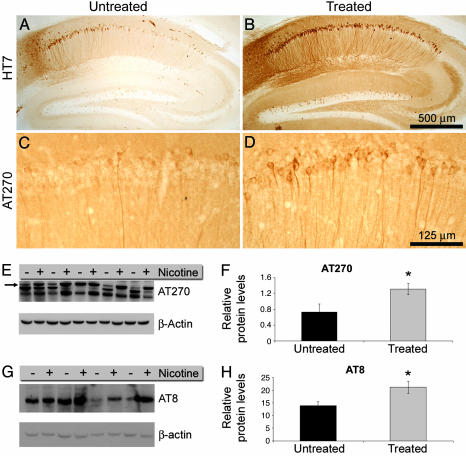
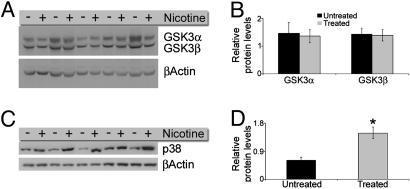
The association between nicotinic acetylcholine receptor (nAChR) dysfunction and cognitive decline in Alzheimer's disease (AD) has been widely exploited for its therapeutic potential. The effects of chronic nicotine exposure on Abeta accumulation have been studied in both humans and animal models, but its therapeutic efficacy for AD neuropathology is still unresolved. To date, no in vivo studies have addressed the consequences of activating nAChRs on tau pathology. To determine the effects of chronic nicotine administration on Abeta and tau pathology, we chronically administrated nicotine to a transgenic model of AD (3xTg-AD) in their drinking water. Here, we show that chronic nicotine intake causes an up-regulation of nicotinic receptors, which correlated with a marked increase in the aggregation and phosphorylation state of tau. These data show that nicotine exacerbates tau pathology in vivo. The increase in tau phosphorylation appears to be due to the activation of p38-mitogen-activated protein kinase, which is known to phosphorylate tau in vivo and in vitro. We also show that the 3xTg-AD mice have an age-dependent reduction of alpha7nAChRs compared with age-matched nontransgenic mice in specific brain regions. The reduction of alpha7nAChRs is first apparent at 6 months of age and is restricted to brain regions that show intraneuronal Abeta(42) accumulation. Finally, this study highlights the importance of testing compounds designed to ameliorate AD pathology in a model with both neuropathological lesions because of the differential effects it can have on either Abeta or tau.
Figures
Similar articles
-
Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer's disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline.J Alzheimers Dis. 2012;30(1):183-208. doi: 10.3233/JAD-2012-111298. J Alzheimers Dis. 2012. PMID: 22406441
-
Effect of huprine X on β-amyloid, synaptophysin and α7 neuronal nicotinic acetylcholine receptors in the brain of 3xTg-AD and APPswe transgenic mice.Neurodegener Dis. 2010;7(6):379-88. doi: 10.1159/000287954. Epub 2010 Aug 4. Neurodegener Dis. 2010. PMID: 20689242
-
Pyruvate prevents the development of age-dependent cognitive deficits in a mouse model of Alzheimer's disease without reducing amyloid and tau pathology.Neurobiol Dis. 2015 Sep;81:214-24. doi: 10.1016/j.nbd.2014.11.013. Epub 2014 Nov 28. Neurobiol Dis. 2015. PMID: 25434488
-
Role of the nicotinic acetylcholine receptor in Alzheimer's disease pathology and treatment.Neuropharmacology. 2015 Sep;96(Pt B):255-62. doi: 10.1016/j.neuropharm.2014.11.018. Epub 2014 Dec 13. Neuropharmacology. 2015. PMID: 25514383 Review.
-
Propagation of Aß pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies.Acta Neuropathol. 2016 Jan;131(1):5-25. doi: 10.1007/s00401-015-1516-y. Epub 2015 Dec 29. Acta Neuropathol. 2016. PMID: 26715565 Review.
Cited by
-
Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer's Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2.Int J Mol Sci. 2023 Dec 18;24(24):17586. doi: 10.3390/ijms242417586. Int J Mol Sci. 2023. PMID: 38139415 Free PMC article.
-
The significance of the cholinergic system in the brain during aging and in Alzheimer's disease.J Neural Transm (Vienna). 2006 Nov;113(11):1625-44. doi: 10.1007/s00702-006-0579-2. Epub 2006 Oct 13. J Neural Transm (Vienna). 2006. PMID: 17039298 Review.
-
Natural products as a source of Alzheimer's drug leads.Nat Prod Rep. 2011 Jan;28(1):48-77. doi: 10.1039/c0np00027b. Epub 2010 Nov 12. Nat Prod Rep. 2011. PMID: 21072430 Free PMC article. Review.
-
Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway.Proc Natl Acad Sci U S A. 2010 Mar 2;107(9):4188-93. doi: 10.1073/pnas.0912263107. Epub 2010 Feb 11. Proc Natl Acad Sci U S A. 2010. PMID: 20150510 Free PMC article.
-
Amyloid beta and the longest-lived rodent: the naked mole-rat as a model for natural protection from Alzheimer's disease.Neurobiol Aging. 2013 Oct;34(10):2352-60. doi: 10.1016/j.neurobiolaging.2013.03.032. Epub 2013 Apr 22. Neurobiol Aging. 2013. PMID: 23618870 Free PMC article.
References
-
- Davies, P. & Maloney, A. J. (1976) Lancet 2, 1403. - PubMed
-
- Muir, J. L. (1997) Pharmacol. Biochem. Behav. 56, 687-696. - PubMed
-
- Woolf, N. J. (1996) Neurobiol. Learn. Mem. 66, 258-266. - PubMed
-
- Wevers, A. & Schroder, H. (1999) J. Alzheimers Dis. 1, 207-219. - PubMed
-
- Wevers, A., Monteggia, L., Nowacki, S., Bloch, W., Schutz, U., Lindstrom, J., Pereira, E. F., Eisenberg, H., Giacobini, E., de Vos, R. A., et al. (1999) Eur. J. Neurosci. 11, 2551-2565. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
