

DIP-chip: rapid and accurate determination of DNA-binding specificity
- PMID: 15710749
- PMCID: PMC551568
- DOI: 10.1101/gr.3256505
DIP-chip: rapid and accurate determination of DNA-binding specificity
Abstract
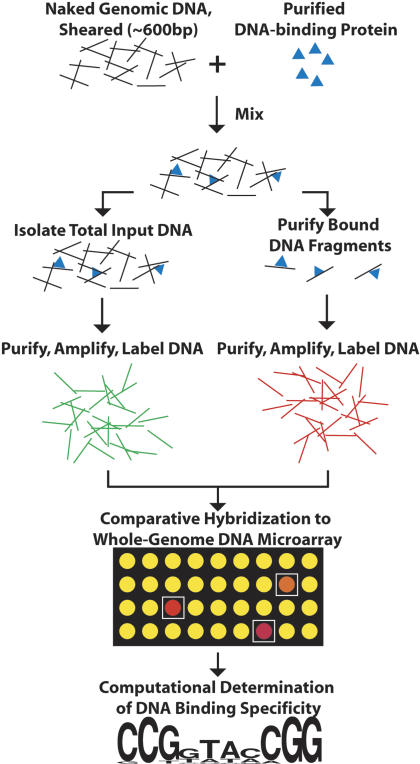
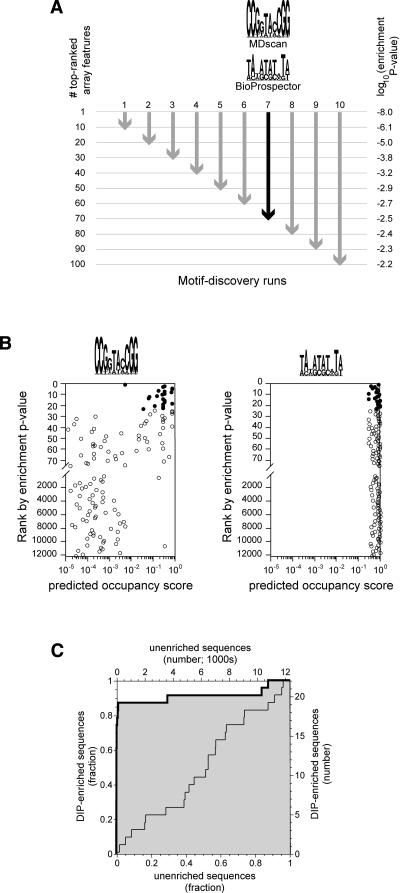
We have developed a new method for determining the DNA-binding specificity of proteins. In DIP-chip (DNA immunoprecipitation with microarray detection), protein.DNA complexes are isolated from an in vitro mixture of purified protein and naked genomic DNA. Whole-genome DNA microarrays are used to identify the protein-bound DNA fragments, and the sequence of the identified fragments is used to derive binding-site descriptions. Using objective criteria for assessing the accuracy of DNA-binding motifs, and using yeast Leu3p as a model, we demonstrate that motifs determined by DIP-chip are as effective at predicting the location of bound proteins in vivo as are motifs determined by conventional low-throughput in vitro methods.
Figures


References
-
- Benjamini, Y. and Hochberg, Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289-300.
-
- Brachmann, C.B., Davies, A., Cost, G.J., Caputo, E., Li, J., Hieter, P., and Boeke, J.D. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115-132. - PubMed
-
- Clarke, N.D. and Granek, J.A. 2003. Rank order metrics for quantifying the association of sequence features with gene regulation. Bioinformatics 19: 212-218. - PubMed
-
- Efron, B. and Gong, G. 1983. A leisurely look at the bootstrap, the jackknife and cross-validation. J. Amer. Stat. Soc. 37: 36-48.
WEB SITE REFERENCES
-
- http://www.bio.unc.edu/faculty/lieb/labpages/Protocols.shtml; Common microarray protocols.
-
- http://www.yeastgenome.org; Saccharomyces Genome Database.
-
- https://genome.unc.edu; UNC Microarray Database.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases