

Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice
- PMID: 15711638
- PMCID: PMC548314
- DOI: 10.1172/JCI22304
Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice
Abstract
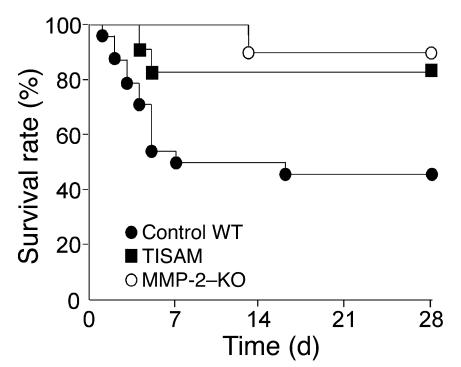
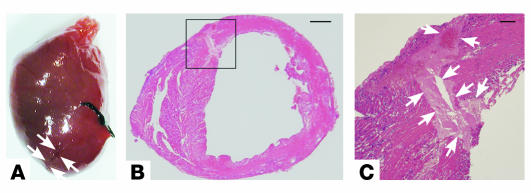
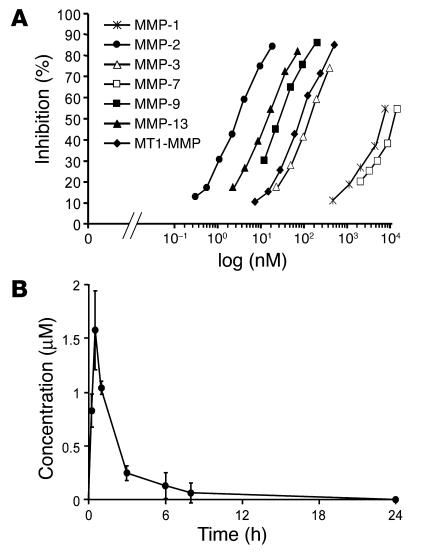
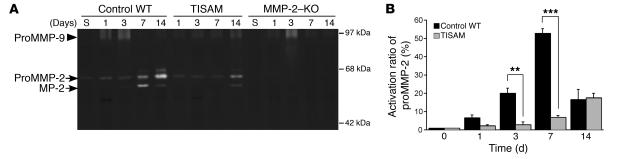
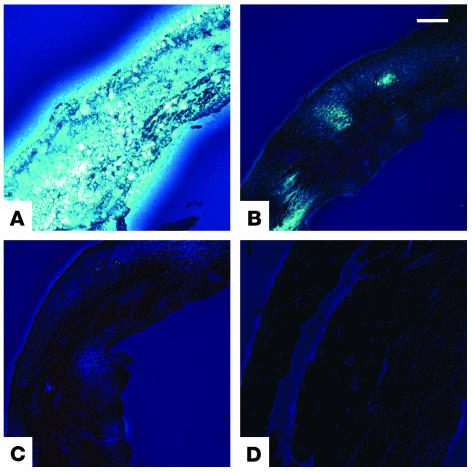
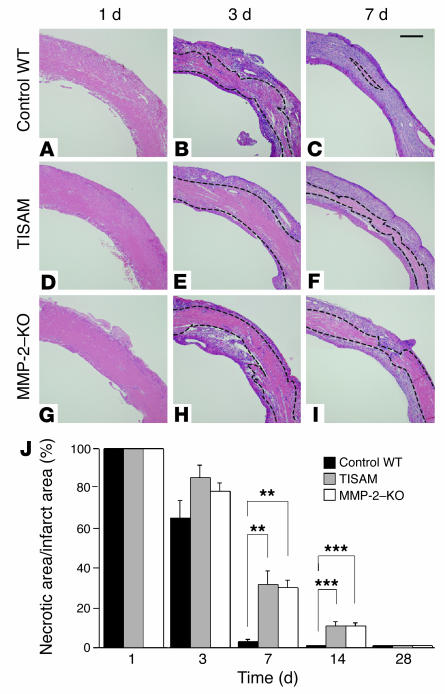
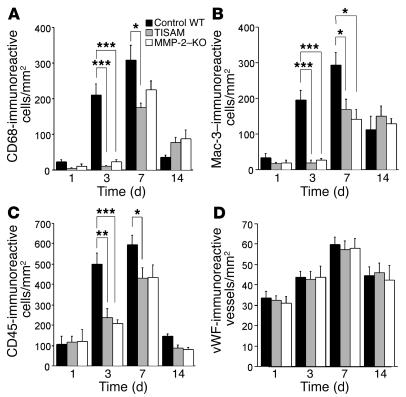
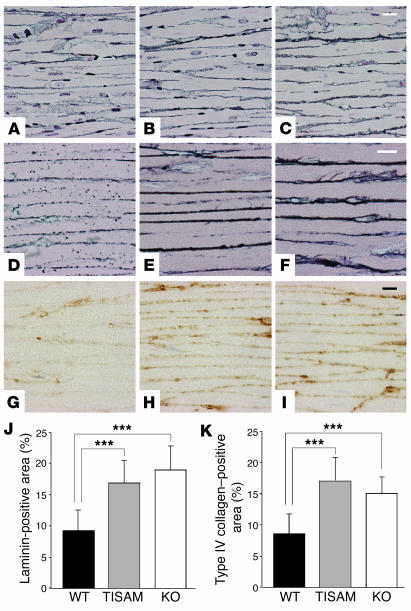
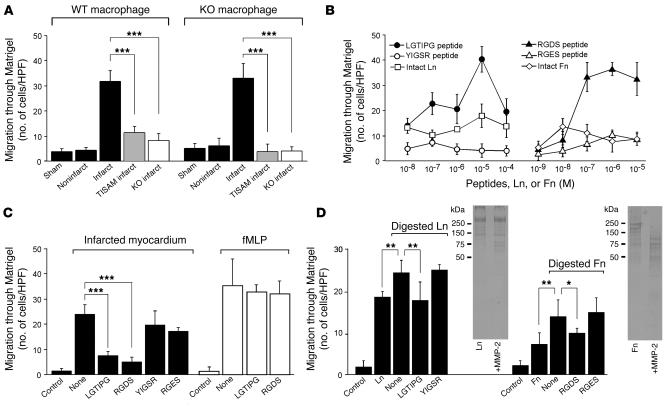
MMPs are implicated in LV remodeling after acute myocardial infarction (MI). To analyze the role of MMP-2, we generated MI by ligating the left coronary artery of MMP-2-KO and WT mice, the latter of which were administered orally an MMP-2-selective inhibitor or vehicle (TISAM). The survival rate was significantly higher in MMP-2-KO and TISAM-treated mice than in control WT mice. The main cause of mortality in control WT mice was cardiac rupture, which was not observed in MMP-2-KO or TISAM-treated mice. Control WT mice, but not MMP-2-KO or TISAM-treated mice, showed activation of the zymogen of MMP-2, strong gelatinolytic activity, and degradation of ECM components, including laminin and fibronectin, in the infarcted myocardium. Although infarcted cardiomyocytes in control WT mice were rapidly removed by macrophages, the removal was suppressed in MMP-2-KO and TISAM-treated mice. Macrophage migration was induced by the infarcted myocardial tissue from control WT mice and was inhibited by treatment of macrophages with laminin or fibronectin peptides prior to migration assay. These data suggest that inhibition of MMP-2 activity improves the survival rate after acute MI by preventing cardiac rupture and delays post-MI remodeling through a reduction in macrophage infiltration.
Figures
References
-
- Antman, E.M., and Braunwald, E. 2001. Acute myocardial infarction. In Harrison’s principles of internal medicine. E. Braunwald, et al., editors. McGraw-Hill. New York, New York, USA. 1386–1399.
-
- Przyklenk K, Connelly CM, McLaughlin RJ, Kloner RA, Apstein CS. Effect of myocyte necrosis on strength, strain, and stiffness of isolated myocardial strips. Am. Heart J. 1987;114:1349–1359. - PubMed
-
- Factor SM, Robinson TF, Dominitz R, Cho SH. Alterations of the myocardial skeletal framework in acute myocardial infarction with and without ventricular rupture. A preliminary report. Am. J. Cardiovasc. Pathol. 1987;1:91–97. - PubMed
-
- Gao XM, et al. Lower risk of postinfarct rupture in mouse heart overexpressing beta 2-adrenergic receptors: importance of collagen content. J. Cardiovasc. Pharmacol. 2002;40:632–640. - PubMed
-
- Ichihara S, et al. Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after acute myocardial infarction. Circulation. 2002;106:2244–2249. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
