

Bcl-2 enhances Ca(2+) signaling to support the intrinsic regenerative capacity of CNS axons
- PMID: 15719013
- PMCID: PMC554135
- DOI: 10.1038/sj.emboj.7600589
Bcl-2 enhances Ca(2+) signaling to support the intrinsic regenerative capacity of CNS axons
Abstract
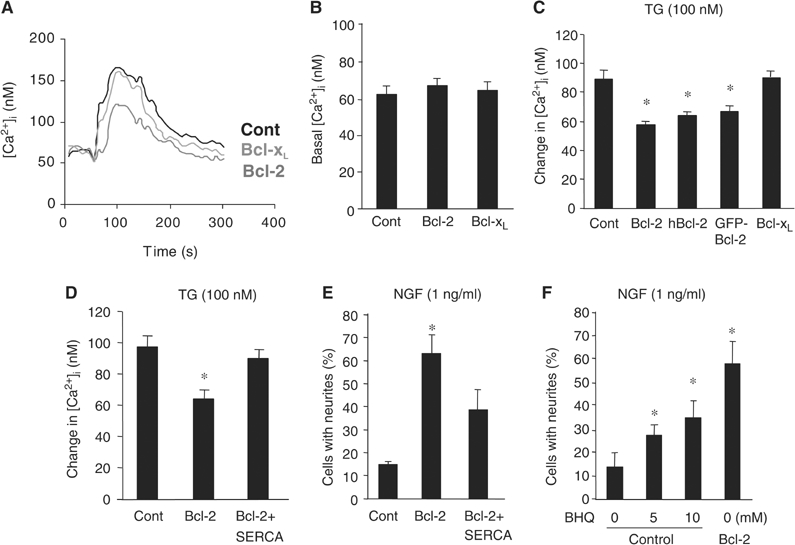
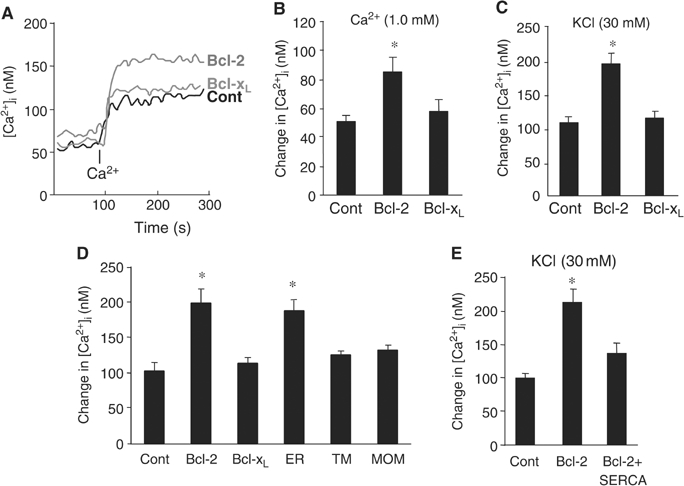
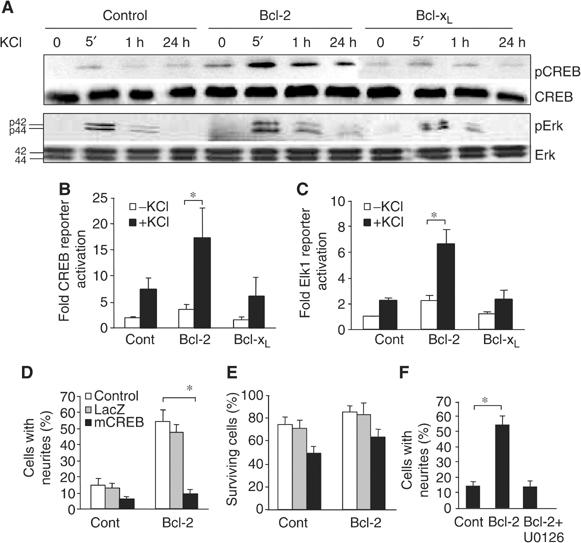
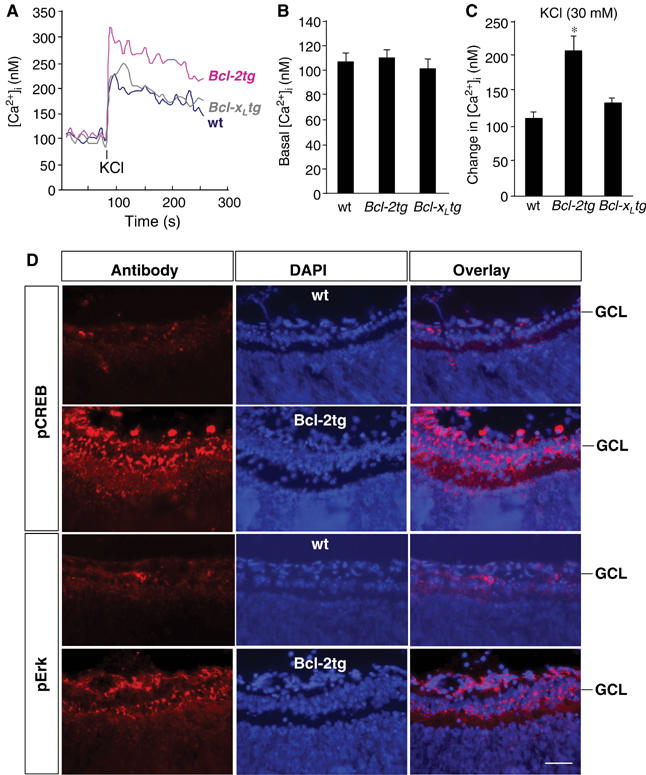
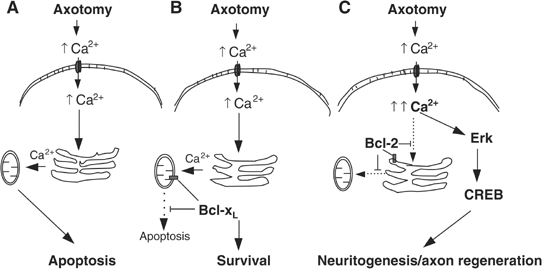
At a certain point in development, axons in the mammalian CNS undergo a profound loss of intrinsic growth capacity, which leads to poor regeneration after injury. Overexpression of Bcl-2 prevents this loss, but the molecular basis of this effect remains unclear. Here, we report that Bcl-2 supports axonal growth by enhancing intracellular Ca(2+) signaling and activating cAMP response element binding protein (CREB) and extracellular-regulated kinase (Erk), which stimulate the regenerative response and neuritogenesis. Expression of Bcl-2 decreases endoplasmic reticulum (ER) Ca(2+) uptake and storage, and thereby leads to a larger intracellular Ca(2+) response induced by Ca(2+) influx or axotomy in Bcl-2-expressing neurons than in control neurons. Bcl-x(L), an antiapoptotic member of the Bcl-2 family that does not affect ER Ca(2+) uptake, supports neuronal survival but cannot activate CREB and Erk or promote axon regeneration. These results suggest a novel role for ER Ca(2+) in the regulation of neuronal response to injury and define a dedicated signaling event through which Bcl-2 supports CNS regeneration.
Figures
Similar articles
-
Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2-dependent mechanism.Invest Ophthalmol Vis Sci. 2003 Jan;44(1):347-54. doi: 10.1167/iovs.02-0198. Invest Ophthalmol Vis Sci. 2003. PMID: 12506095
-
Fibroblast growth factor 2 applied to the optic nerve after axotomy increases Bcl-2 and decreases Bax in ganglion cells by activating the extracellular signal-regulated kinase signaling pathway.J Neurochem. 2005 Jun;93(6):1422-33. doi: 10.1111/j.1471-4159.2005.03129.x. J Neurochem. 2005. PMID: 15935058
-
[Optic nerve regeneration in Bcl-2 overexpressing mice].Zhonghua Yan Ke Za Zhi. 2005 Sep;41(9):832-6. Zhonghua Yan Ke Za Zhi. 2005. PMID: 16191352 Chinese.
-
Neural development: axon regeneration derailed by dendrites.Curr Biol. 2002 Jul 9;12(13):R455-7. doi: 10.1016/s0960-9822(02)00944-2. Curr Biol. 2002. PMID: 12121636 Review.
-
Stimulating axonal regeneration of mature retinal ganglion cells and overcoming inhibitory signaling.Cell Tissue Res. 2012 Jul;349(1):79-85. doi: 10.1007/s00441-011-1302-7. Cell Tissue Res. 2012. PMID: 22293973 Review.
Cited by
-
Acute leptin treatment enhances functional recovery after spinal cord injury.PLoS One. 2012;7(4):e35594. doi: 10.1371/journal.pone.0035594. Epub 2012 Apr 20. PLoS One. 2012. PMID: 22536415 Free PMC article.
-
Re-establishing the regenerative potential of central nervous system axons in postnatal mice.J Cell Sci. 2005 Mar 1;118(Pt 5):863-72. doi: 10.1242/jcs.01658. J Cell Sci. 2005. PMID: 15731004 Free PMC article.
-
MAPK/ERK Pathway as a Central Regulator in Vertebrate Organ Regeneration.Int J Mol Sci. 2022 Jan 27;23(3):1464. doi: 10.3390/ijms23031464. Int J Mol Sci. 2022. PMID: 35163418 Free PMC article. Review.
-
Emerging roles for insulin-like growth factor binding protein like protein 1.Neural Regen Res. 2019 Feb;14(2):258-259. doi: 10.4103/1673-5374.244787. Neural Regen Res. 2019. PMID: 30531006 Free PMC article. No abstract available.
-
Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders.Antioxid Redox Signal. 2010 Dec 1;13(11):1763-811. doi: 10.1089/ars.2009.3074. Epub 2010 Aug 28. Antioxid Redox Signal. 2010. PMID: 20446769 Free PMC article. Review.
References
-
- Adams JP, Sweatt JD (2002) Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 42: 135–163 - PubMed
-
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13–26 - PubMed
-
- Chen DF, Schneider GE, Martinou JC, Tonegawa S (1997) Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385: 434–439 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
