

Identification of a new family of putative PD-(D/E)XK nucleases with unusual phylogenomic distribution and a new type of the active site
- PMID: 15720711
- PMCID: PMC551604
- DOI: 10.1186/1471-2164-6-21
Identification of a new family of putative PD-(D/E)XK nucleases with unusual phylogenomic distribution and a new type of the active site
Abstract
Background: Prediction of structure and function for uncharacterized protein families by identification of evolutionary links to characterized families and known structures is one of the cornerstones of genomics. Theoretical assignment of three-dimensional folds and prediction of protein function even at a very general level can facilitate the experimental determination of the molecular mechanism of action and the role that members of a given protein family fulfill in the cell. Here, we predict the three-dimensional fold and study the phylogenomic distribution of members of a large family of uncharacterized proteins classified in the Clusters of Orthologous Groups database as COG4636.
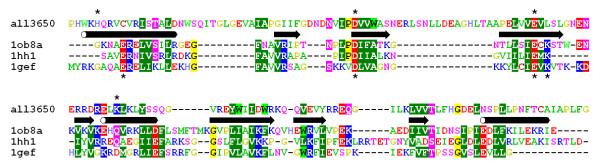
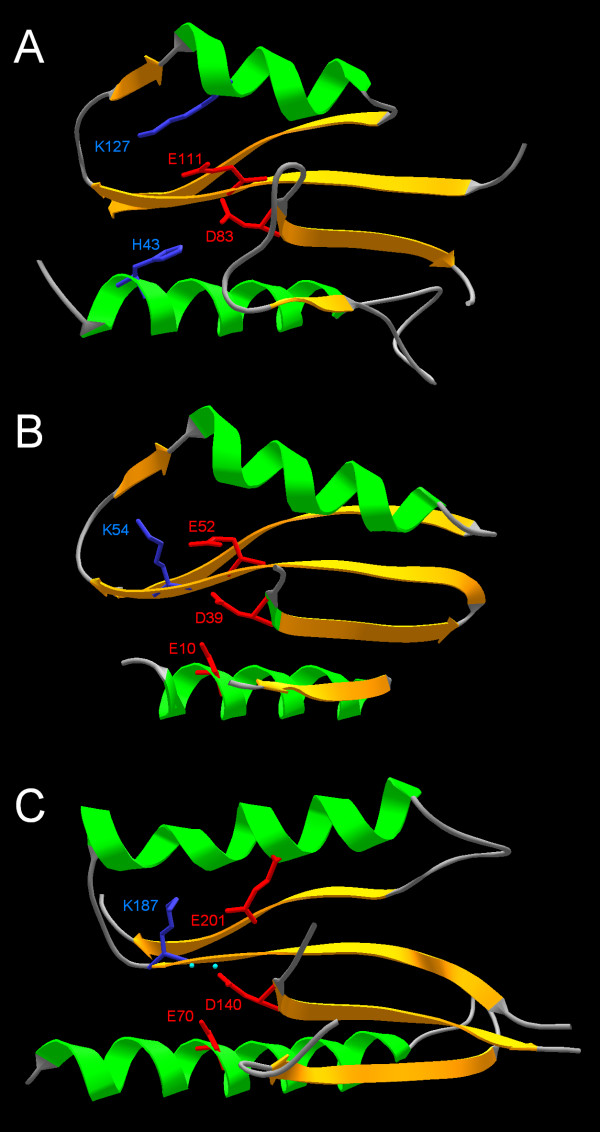
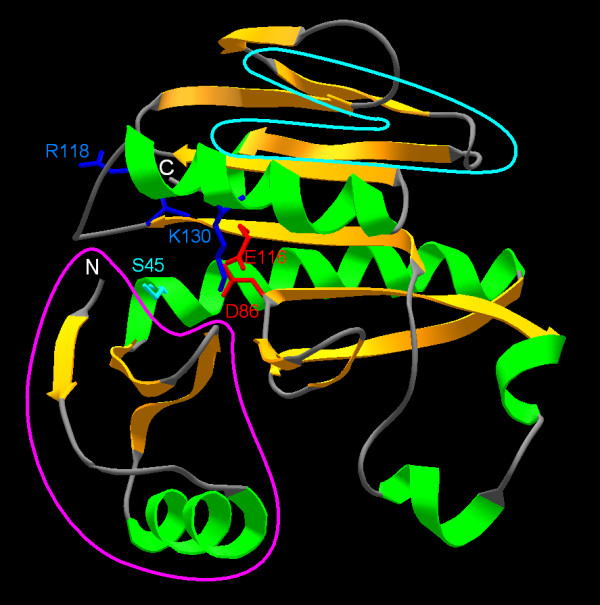
Results: Using protein fold-recognition we found that members of COG4636 are remotely related to Holliday junction resolvases and other nucleases from the PD-(D/E)XK superfamily. Structure modeling and sequence analyses suggest that most members of COG4636 exhibit a new, unusual variant of the putative active site, in which the catalytic Lys residue migrated in the sequence, but retained similar spatial position with respect to other functionally important residues. Sequence analyses revealed that members of COG4636 and their homologs are found mainly in Cyanobacteria, but also in other bacterial phyla. They undergo horizontal transfer and extensive proliferation in the colonized genomes; for instance in Gloeobacter violaceus PCC 7421 they comprise over 2% of all protein-encoding genes. Thus, members of COG4636 appear to be a new type of selfish genetic elements, which may fulfill an important role in the genome dynamics of Cyanobacteria and other species they invaded. Our analyses provide a platform for experimental determination of the molecular and cellular function of members of this large protein family.
Conclusion: After submission of this manuscript, a crystal structure of one of the COG4636 members was released in the Protein Data Bank (code 1wdj; Idaka, M., Wada, T., Murayama, K., Terada, T., Kuramitsu, S., Shirouzu, M., Yokoyama, S.: Crystal structure of Tt1808 from Thermus thermophilus Hb8, to be published). Our analysis of the Tt1808 structure reveals that we correctly predicted all functionally important features of the COG4636 family, including the membership in the PD-(D/E)xK superfamily of nucleases, the three-dimensional fold, the putative catalytic residues, and the unusual configuration of the active site.
Figures



Similar articles
-
Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile-profile alignments.Nucleic Acids Res. 2011 Mar;39(4):1187-96. doi: 10.1093/nar/gkq958. Epub 2010 Oct 20. Nucleic Acids Res. 2011. PMID: 20961958 Free PMC article.
-
The PD-(D/E)XK superfamily revisited: identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function.BMC Bioinformatics. 2005 Jul 12;6:172. doi: 10.1186/1471-2105-6-172. BMC Bioinformatics. 2005. PMID: 16011798 Free PMC article.
-
Realm of PD-(D/E)XK nuclease superfamily revisited: detection of novel families with modified transitive meta profile searches.BMC Struct Biol. 2007 Jun 20;7:40. doi: 10.1186/1472-6807-7-40. BMC Struct Biol. 2007. PMID: 17584917 Free PMC article.
-
Prediction of protein function from protein sequence and structure.Q Rev Biophys. 2003 Aug;36(3):307-40. doi: 10.1017/s0033583503003901. Q Rev Biophys. 2003. PMID: 15029827 Review.
-
Structure and mechanism of nucleases regulated by SLX4.Curr Opin Struct Biol. 2016 Feb;36:97-105. doi: 10.1016/j.sbi.2016.01.003. Epub 2016 Jan 29. Curr Opin Struct Biol. 2016. PMID: 26827285 Review.
Cited by
-
Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile-profile alignments.Nucleic Acids Res. 2011 Mar;39(4):1187-96. doi: 10.1093/nar/gkq958. Epub 2010 Oct 20. Nucleic Acids Res. 2011. PMID: 20961958 Free PMC article.
-
Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily.Nucleic Acids Res. 2012 Aug;40(15):7016-45. doi: 10.1093/nar/gks382. Epub 2012 May 25. Nucleic Acids Res. 2012. PMID: 22638584 Free PMC article.
-
Cloning and analysis of a bifunctional methyltransferase/restriction endonuclease TspGWI, the prototype of a Thermus sp. enzyme family.BMC Mol Biol. 2009 May 29;10:52. doi: 10.1186/1471-2199-10-52. BMC Mol Biol. 2009. PMID: 19480701 Free PMC article.
-
The PD-(D/E)XK superfamily revisited: identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function.BMC Bioinformatics. 2005 Jul 12;6:172. doi: 10.1186/1471-2105-6-172. BMC Bioinformatics. 2005. PMID: 16011798 Free PMC article.
-
HsdR subunit of the type I restriction-modification enzyme EcoR124I: biophysical characterisation and structural modelling.J Mol Biol. 2008 Feb 15;376(2):438-452. doi: 10.1016/j.jmb.2007.11.024. Epub 2007 Nov 17. J Mol Biol. 2008. PMID: 18164032 Free PMC article.
References
-
- Aggarwal AK. Structure and function of restriction endonucleases. CurrOpinStructBiol. 1995;5:11–19. - PubMed
-
- Bujnicki JM. Molecular phylogenetics of restriction endonucleases. In: Pingoud A, editor. Restriction Endonucleases. Vol. 14. Berlin, Springer-Verlag; 2004. pp. 63–87. (Nucleic Acids and Molecular Biology). Gross HJ.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
