

Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release
- PMID: 15730564
- PMCID: PMC555946
- DOI: 10.1186/1471-2202-6-13
Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release
Abstract
Background: Apoptosis plays a key role in cell death observed in neurodegenerative diseases marked by a progressive loss of neurons as seen in Alzheimer's disease. Although the exact cause of apoptosis is not known, a number of factors such as free radicals, insufficient levels of nerve growth factors and excessive levels of glutamate have been implicated. We and others, have previously reported that in a stable HT22 neuronal cell line, glutamate induces apoptosis as indicated by DNA fragmentation and up- and down-regulation of Bax (pro-apoptotic), and Bcl-2 (anti-apoptotic) genes respectively. Furthermore, these changes were reversed/inhibited by estrogens. Several lines of evidence also indicate that a family of cysteine proteases (caspases) appear to play a critical role in neuronal apoptosis. The purpose of the present study is to determine in primary cultures of cortical cells, if glutamate-induced neuronal apoptosis and its inhibition by estrogens involve changes in caspase-3 protease and whether this process is mediated by Fas receptor and/or mitochondrial signal transduction pathways involving release of cytochrome c.
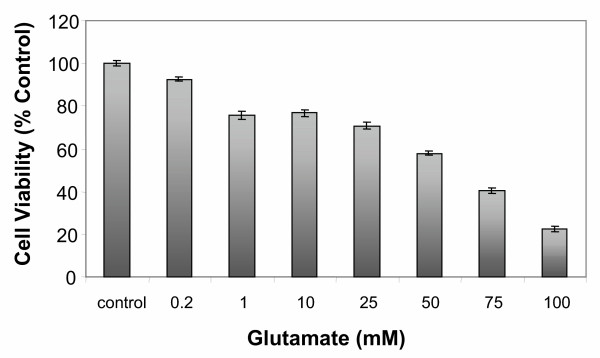
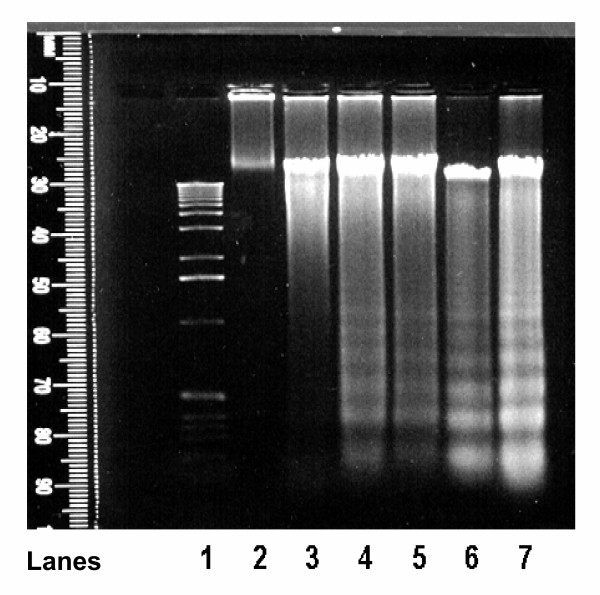
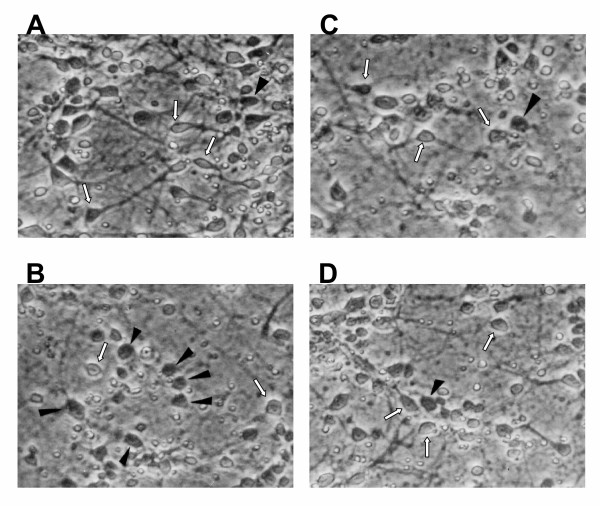
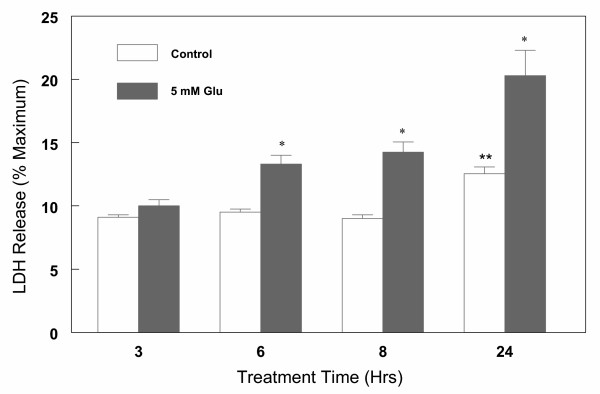
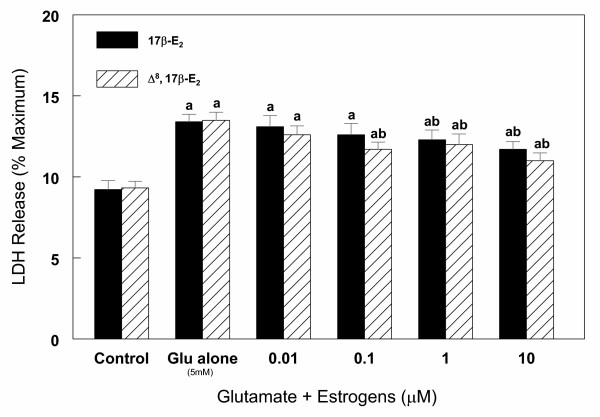
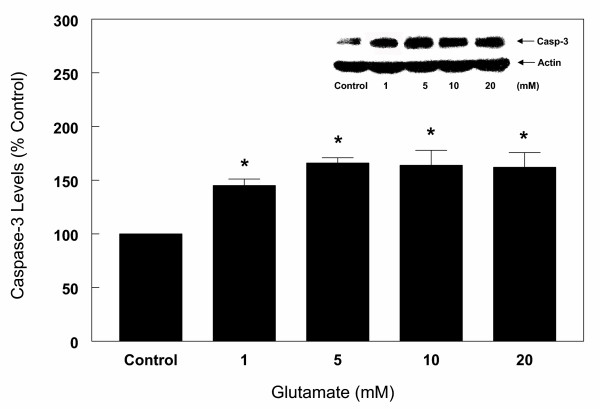
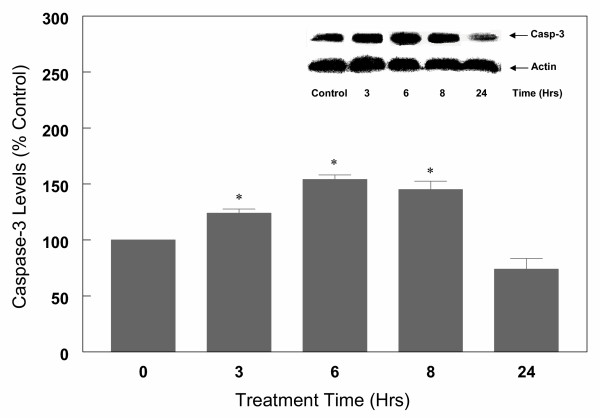
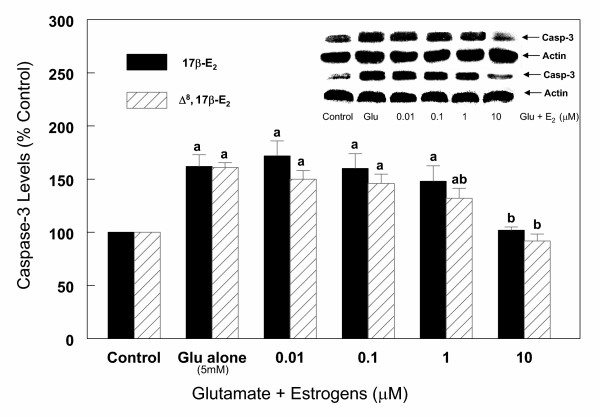
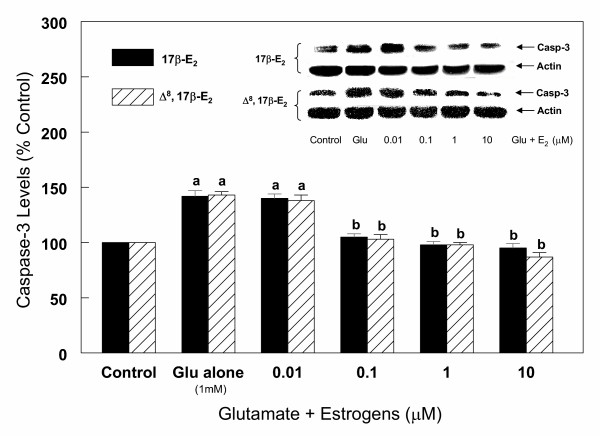
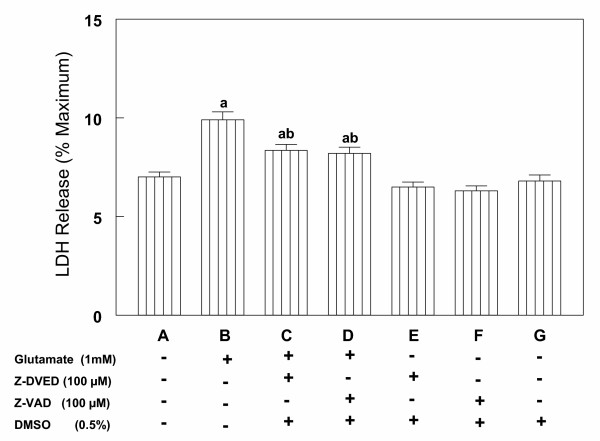
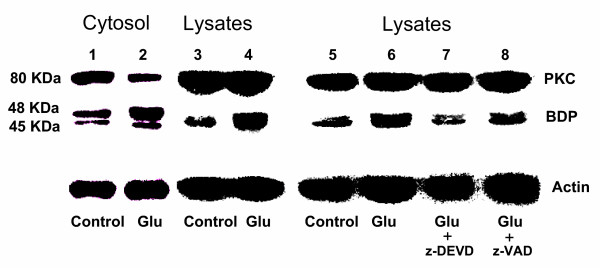
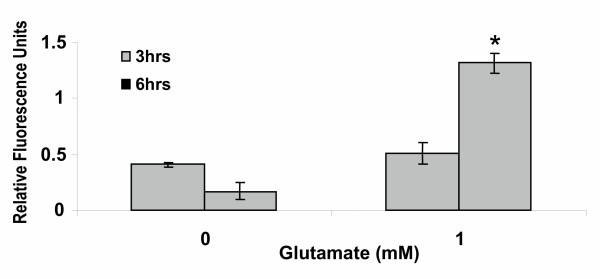
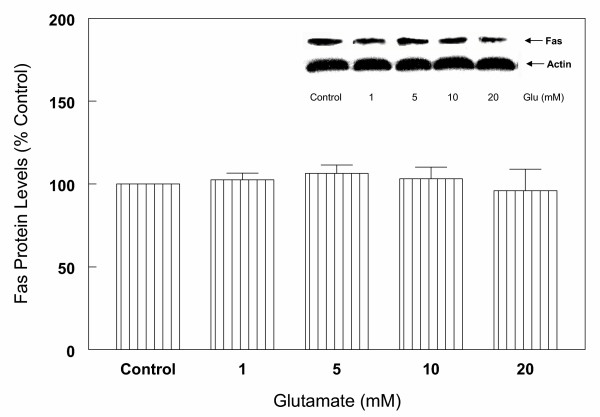
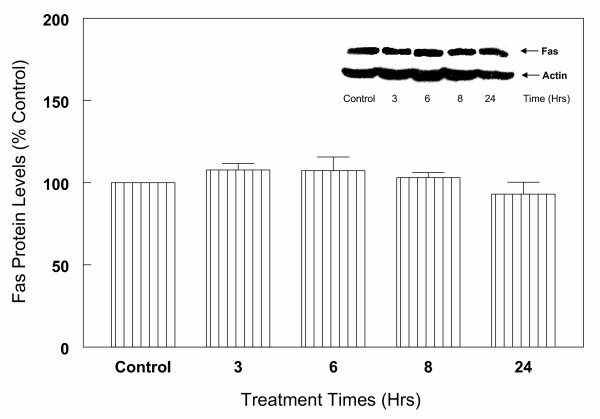
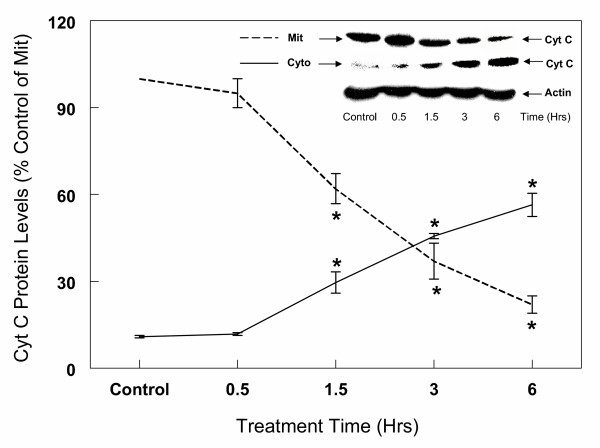
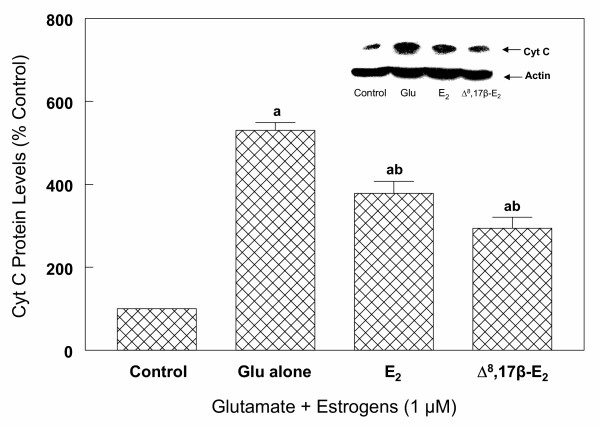
Results: In primary cultures of rat cortical cells, glutamate induced apoptosis that was associated with enhanced DNA fragmentation, morphological changes, and up-regulation of pro-caspase-3. Exposure of cortical cells to glutamate resulted in a time-dependent cell death and an increase in caspase-3 protein levels. Although the increase in caspase-3 levels was evident after 3 h, cell death was only significantly increased after 6 h. Treatment of cells for 6 h with 1 to 20 mM glutamate resulted in a 35 to 45% cell death that was associated with a 45 to 65% increase in the expression of caspase-3 protein. Pretreatment with caspase-3-protease inhibitor z-DEVD or pan-caspase inhibitor z-VAD significantly decreased glutamate-induced cell death of cortical cells. Exposure of cells to glutamate for 6 h in the presence or absence of 17beta-estradiol or Delta8, 17beta-estradiol (10 nM-10 microM) resulted in the prevention of cell death and was associated with a significant dose-dependent decrease in caspase-3 protein levels, with Delta8, 17beta-E2 being more potent than 17beta-E2. Protein levels of Fas receptor remained unchanged in the presence of glutamate. In contrast, treatment with glutamate induced, in a time-dependent manner, the release of cytochrome c into the cytosol. Cytosolic cytochrome c increased as early as 1.5 h after glutamate treatment and these levels were 5 fold higher after 6 h, compared to levels in the untreated cells. Concomitant with these changes, the levels of cytochrome c in mitochondria decreased significantly. Both 17beta-E2 and Delta8, 17beta-E2 reduced the release of cytochrome c from mitochondria into the cytosol and this decrease in cytosolic cytochrome c was associated with inhibition of glutamate-induced cell death.
Conclusion: In the primary cortical cells, glutamate-induced apoptosis is accompanied by up-regulation of caspase-3 and its activity is blocked by caspase protease inhibitors. These effects of glutamate on caspase-3 appear to be independent of changes in Fas receptor, but are associated with the rapid release of mitochondrial cytochrome c, which precedes changes in caspase-3 protein levels leading to apoptotic cell death. This process was differentially inhibited by estrogens with the novel equine estrogen Delta8, 17beta-E2 being more potent than 17beta-E2. To our knowledge, this is the first study to demonstrate that equine estrogens can prevent glutamate-induced translocation of cytochrome c from mitochondria to cytosol in rat primary cortical cells.
Figures
References
-
- Fonnum F. Glutamate: a neurotransmitter in mammalia brain. J Neurochem. 1984;42:1–11. - PubMed
-
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. - PubMed
-
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
