

SARS transmission pattern in Singapore reassessed by viral sequence variation analysis
- PMID: 15736999
- PMCID: PMC549591
- DOI: 10.1371/journal.pmed.0020043
SARS transmission pattern in Singapore reassessed by viral sequence variation analysis
Abstract
Background: Epidemiological investigations of infectious disease are mainly dependent on indirect contact information and only occasionally assisted by characterization of pathogen sequence variation from clinical isolates. Direct sequence analysis of the pathogen, particularly at a population level, is generally thought to be too cumbersome, technically difficult, and expensive. We present here a novel application of mass spectrometry (MS)-based technology in characterizing viral sequence variations that overcomes these problems, and we apply it retrospectively to the severe acute respiratory syndrome (SARS) outbreak in Singapore.
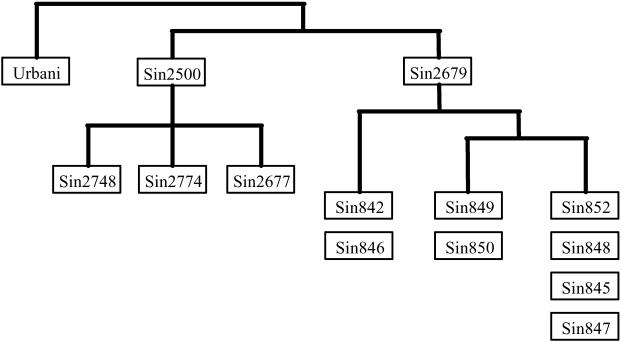
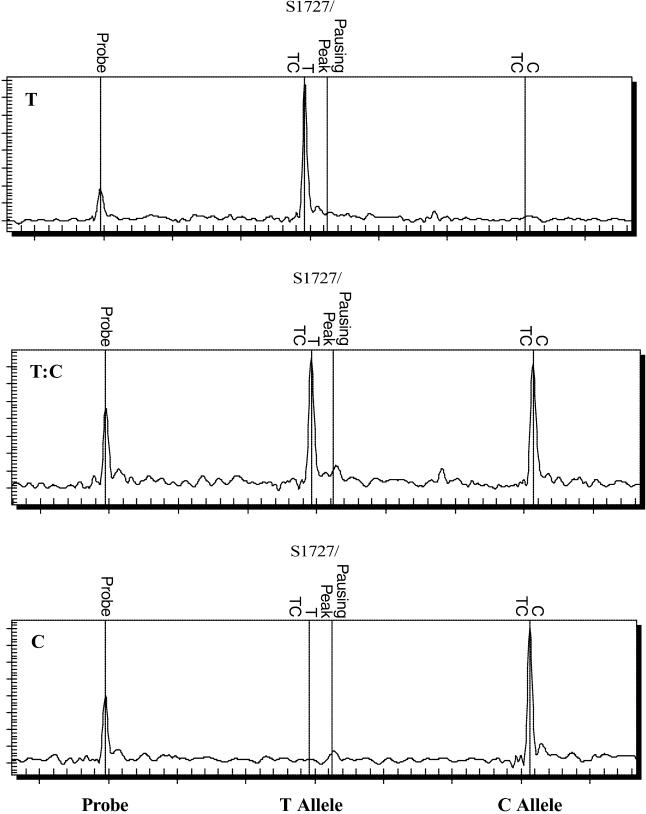
Methods and findings: The success rate of the MS-based analysis for detecting SARS coronavirus (SARS-CoV) sequence variations was determined to be 95% with 75 copies of viral RNA per reaction, which is sufficient to directly analyze both clinical and cultured samples. Analysis of 13 SARS-CoV isolates from the different stages of the Singapore outbreak identified nine sequence variations that could define the molecular relationship between them and pointed to a new, previously unidentified, primary route of introduction of SARS-CoV into the Singapore population. Our direct determination of viral sequence variation from a clinical sample also clarified an unresolved epidemiological link regarding the acquisition of SARS in a German patient. We were also able to detect heterogeneous viral sequences in primary lung tissues, suggesting a possible coevolution of quasispecies of virus within a single host.
Conclusion: This study has further demonstrated the importance of improving clinical and epidemiological studies of pathogen transmission through the use of genetic analysis and has revealed the MS-based analysis to be a sensitive and accurate method for characterizing SARS-CoV genetic variations in clinical samples. We suggest that this approach should be used routinely during outbreaks of a wide variety of agents, in order to allow the most effective control.
Conflict of interest statement
ETL is a member of the Editorial Board of
Figures
References
-
- Peiris JS, Kwok YY, Osterhaus AD, Stöhr K. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431–2441. - PubMed
-
- Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. - PubMed
-
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1398. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
