

Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy
- PMID: 15737406
- PMCID: PMC2819430
- DOI: 10.1016/j.pharmthera.2004.10.004
Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy
Erratum in
- Pharmacol Ther. 2006 Jul;111(1):287
Corrected and republished in
-
Erratum to "Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintenance of epilepsy." [Pharmacol. Ther. 105(3) (2005) 229-266].Pharmacol Ther. 2006 Jul;111(1):288-325. doi: 10.1016/j.pharmthera.2004.10.015. Pharmacol Ther. 2006. PMID: 16832874 Review.
Abstract
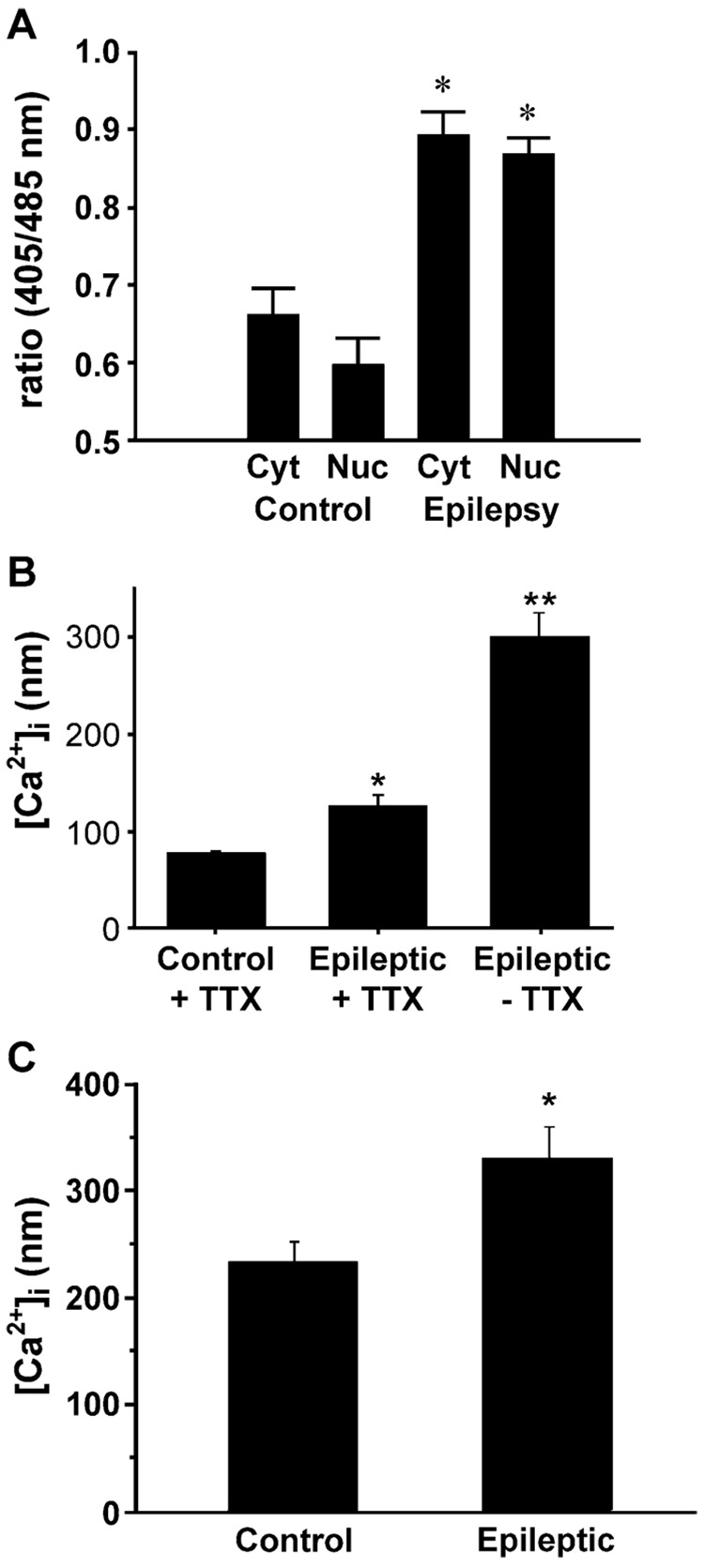
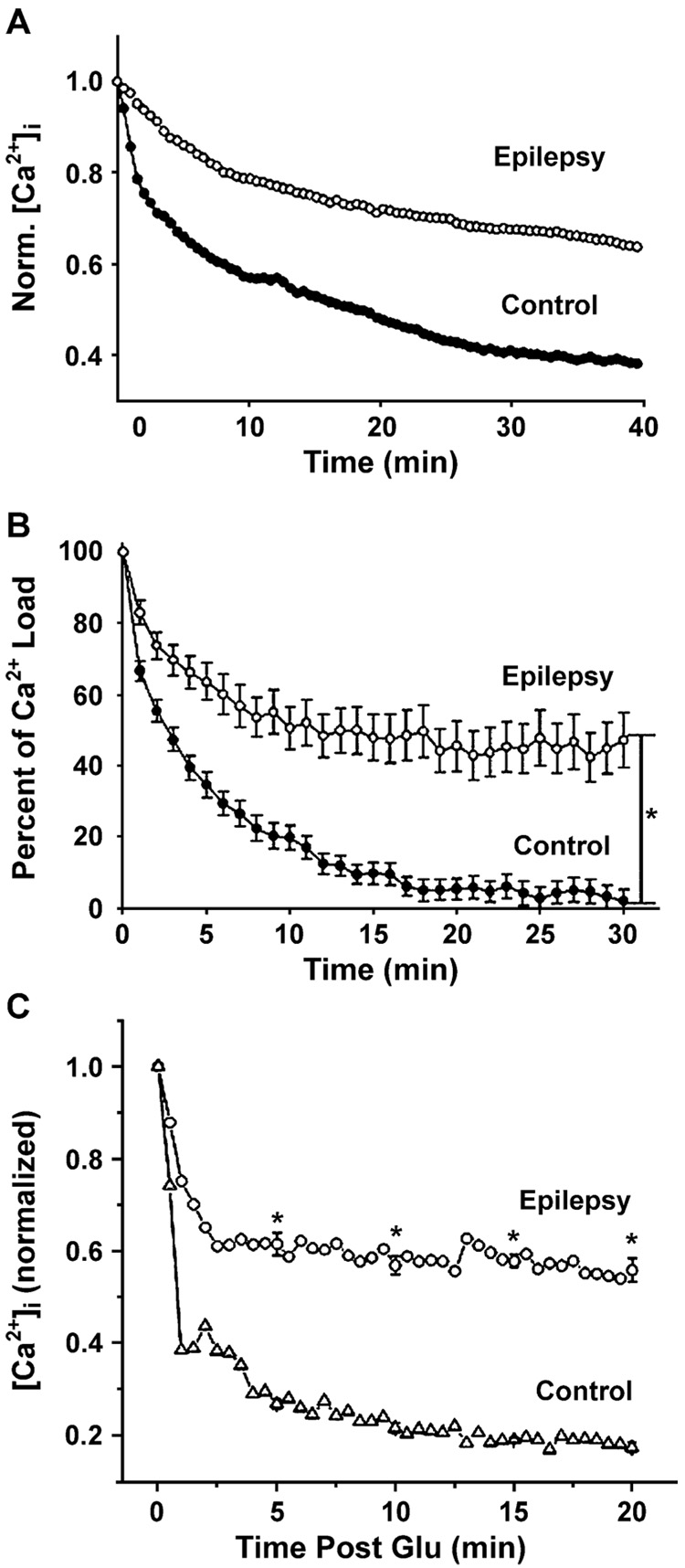
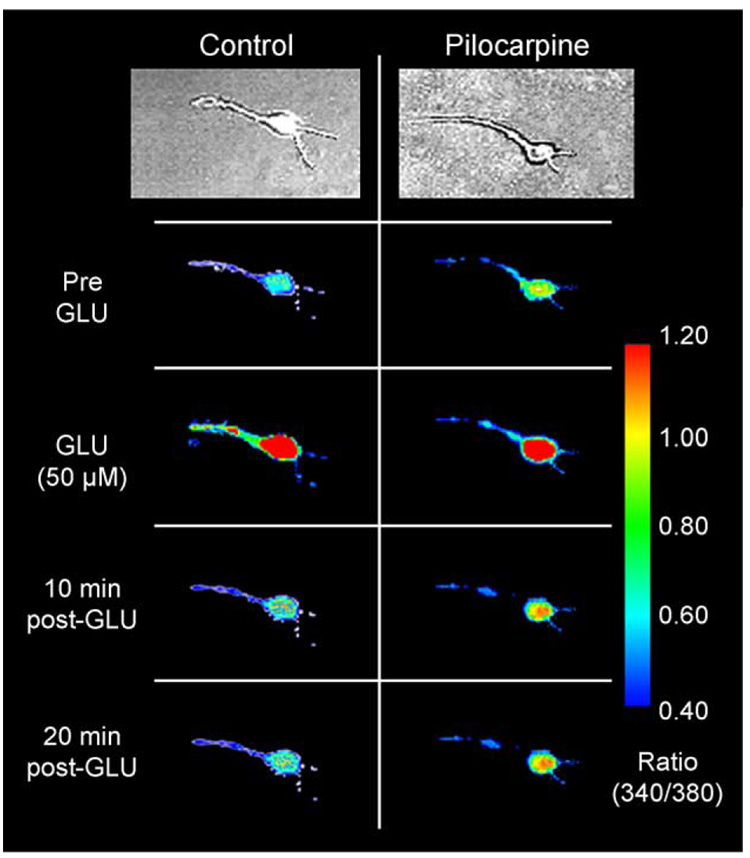
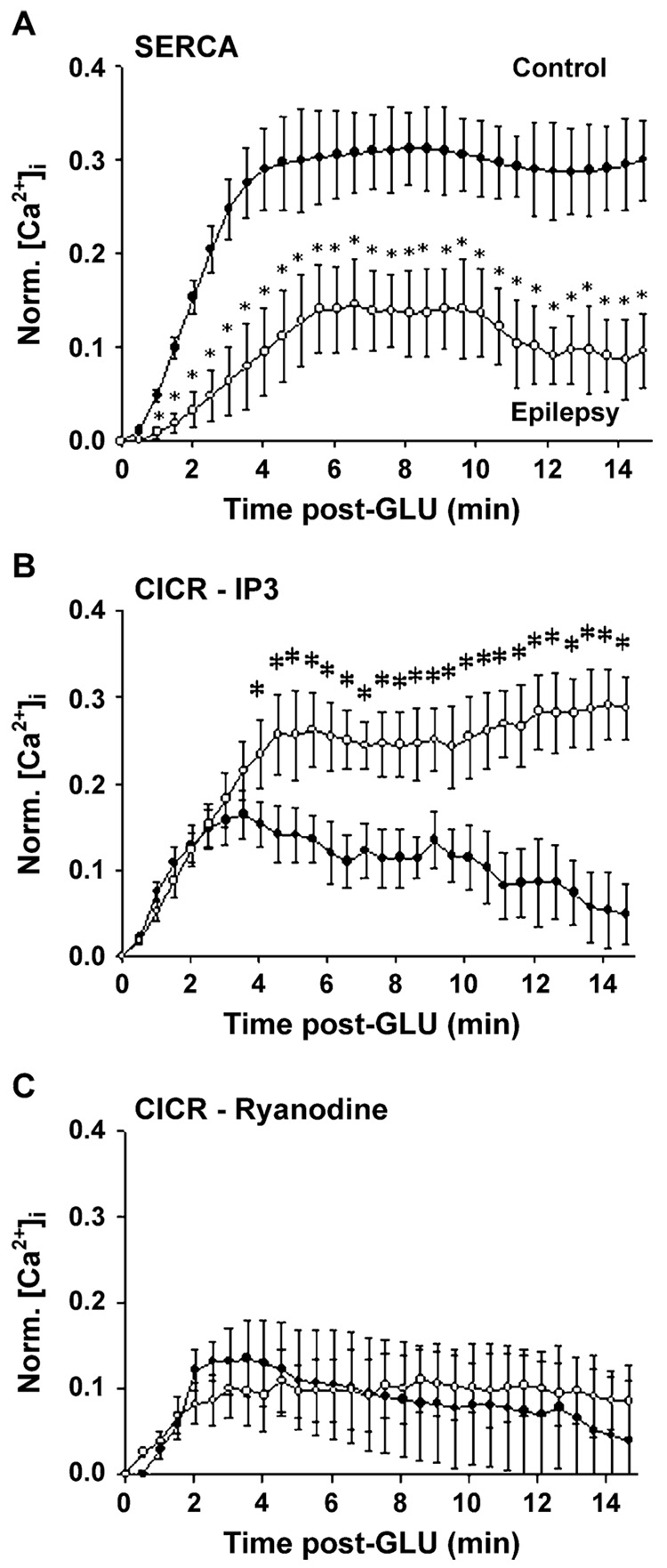
Epilepsy is one of the most common neurological disorders. Although epilepsy can be idiopathic, it is estimated that up to 50% of all epilepsy cases are initiated by neurological insults and are called acquired epilepsy (AE). AE develops in 3 phases: (1) the injury (central nervous system [CNS] insult), (2) epileptogenesis (latency), and (3) the chronic epileptic (spontaneous recurrent seizure) phases. Status epilepticus (SE), stroke, and traumatic brain injury (TBI) are 3 major examples of common brain injuries that can lead to the development of AE. It is especially important to understand the molecular mechanisms that cause AE because it may lead to innovative strategies to prevent or cure this common condition. Recent studies have offered new insights into the cause of AE and indicate that injury-induced alterations in intracellular calcium concentration levels [Ca(2+)](i) and calcium homeostatic mechanisms play a role in the development and maintenance of AE. The injuries that cause AE are different, but they share a common molecular mechanism for producing brain damage-an increase in extracellular glutamate concentration that causes increased intracellular neuronal calcium, leading to neuronal injury and/or death. Neurons that survive the injury induced by glutamate and are exposed to increased [Ca(2+)](i) are the cellular substrates to develop epilepsy because dead cells do not seize. The neurons that survive injury sustain permanent long-term plasticity changes in [Ca(2+)](i) and calcium homeostatic mechanisms that are permanent and are a prominent feature of the epileptic phenotype. In the last several years, evidence has accumulated indicating that the prolonged alteration in neuronal calcium dynamics plays an important role in the induction and maintenance of the prolonged neuroplasticity changes underlying the epileptic phenotype. Understanding the role of calcium as a second messenger in the induction and maintenance of epilepsy may provide novel insights into therapeutic advances that will prevent and even cure AE.
Figures
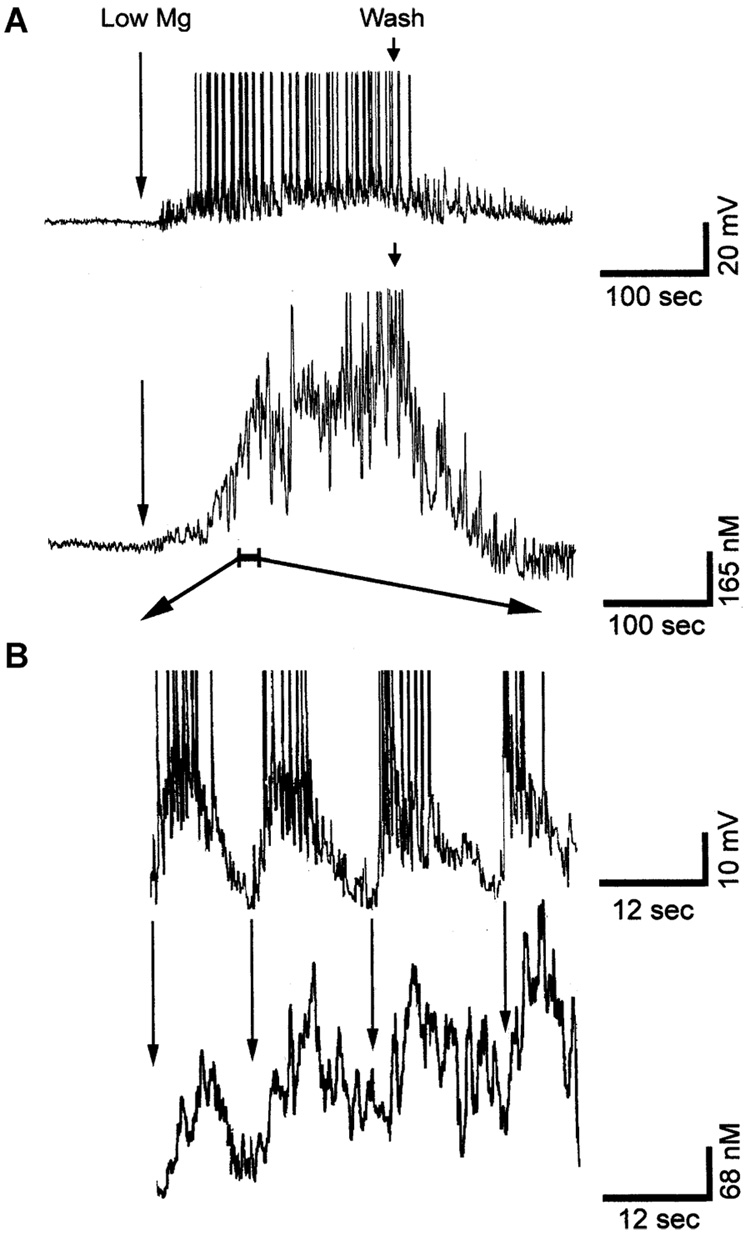
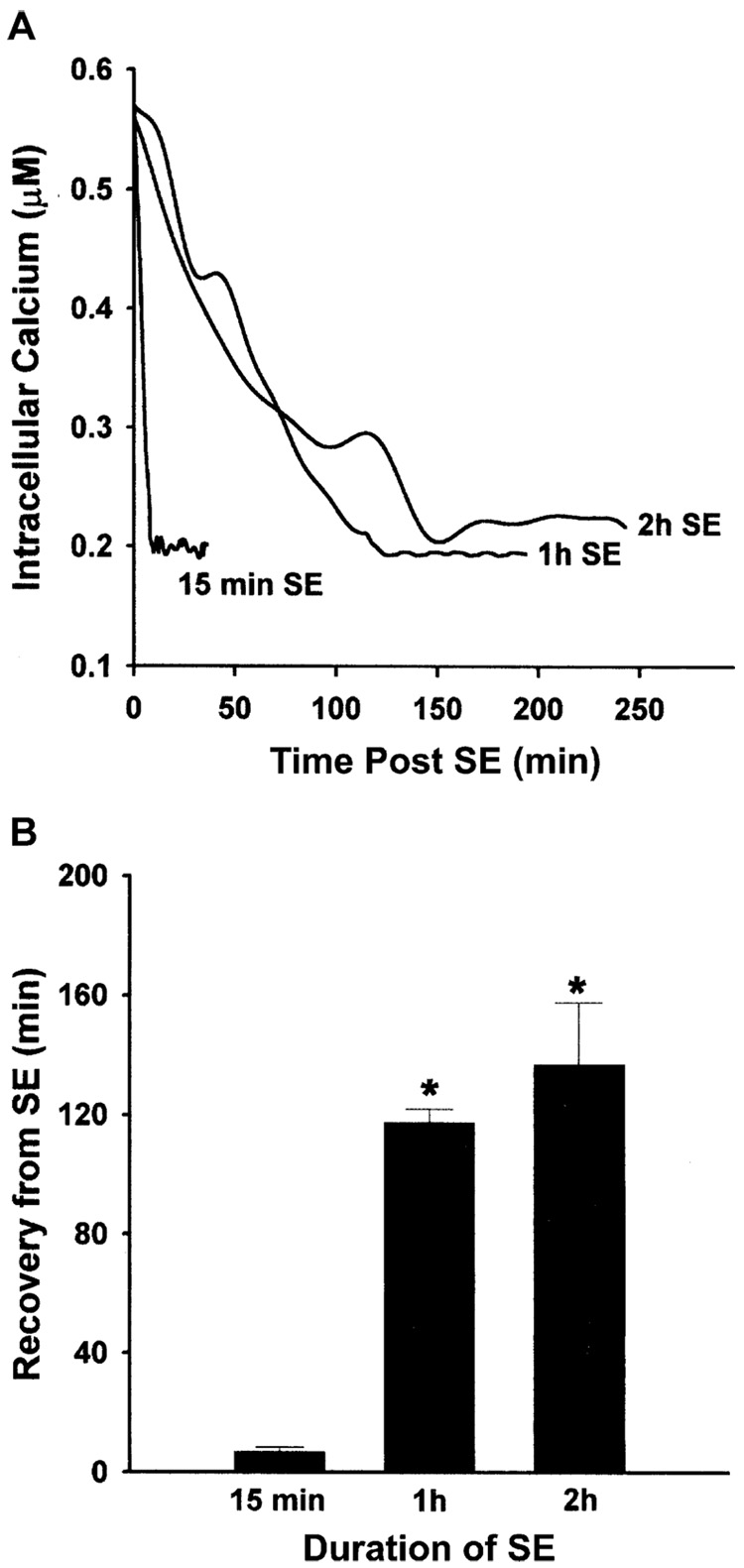
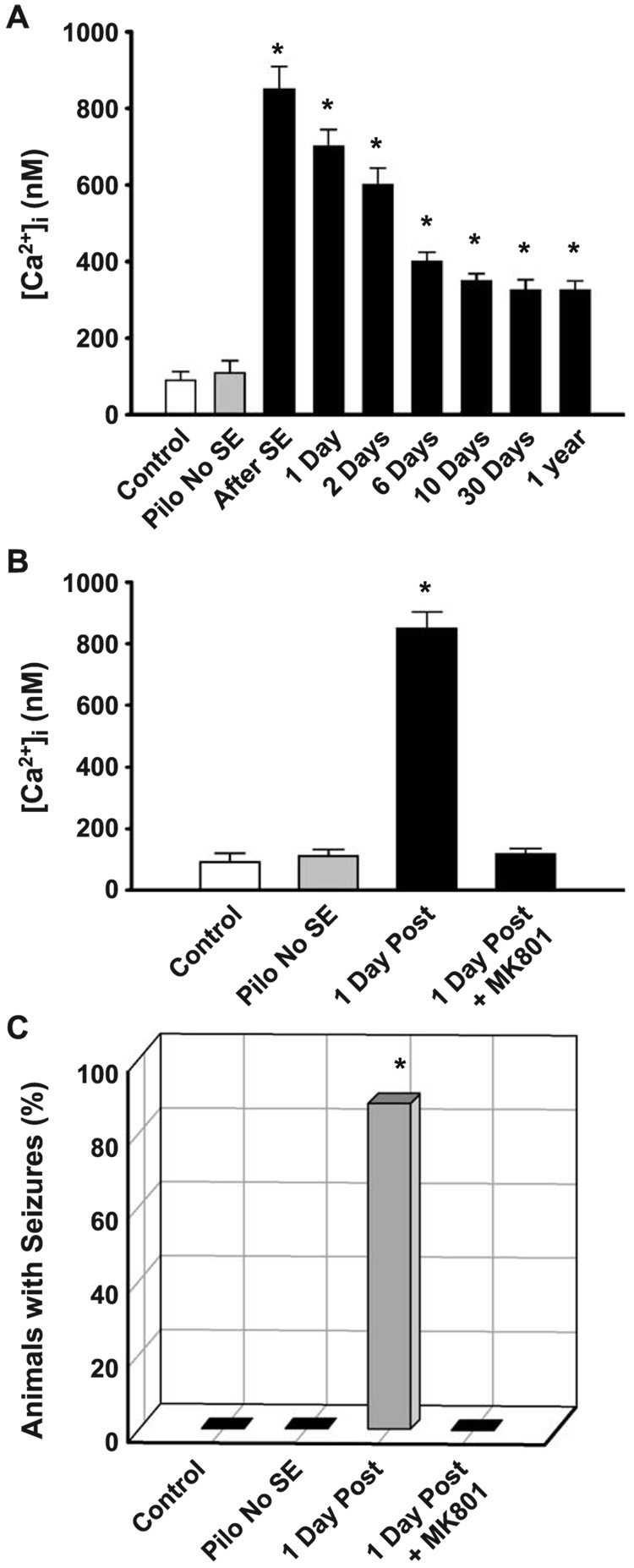
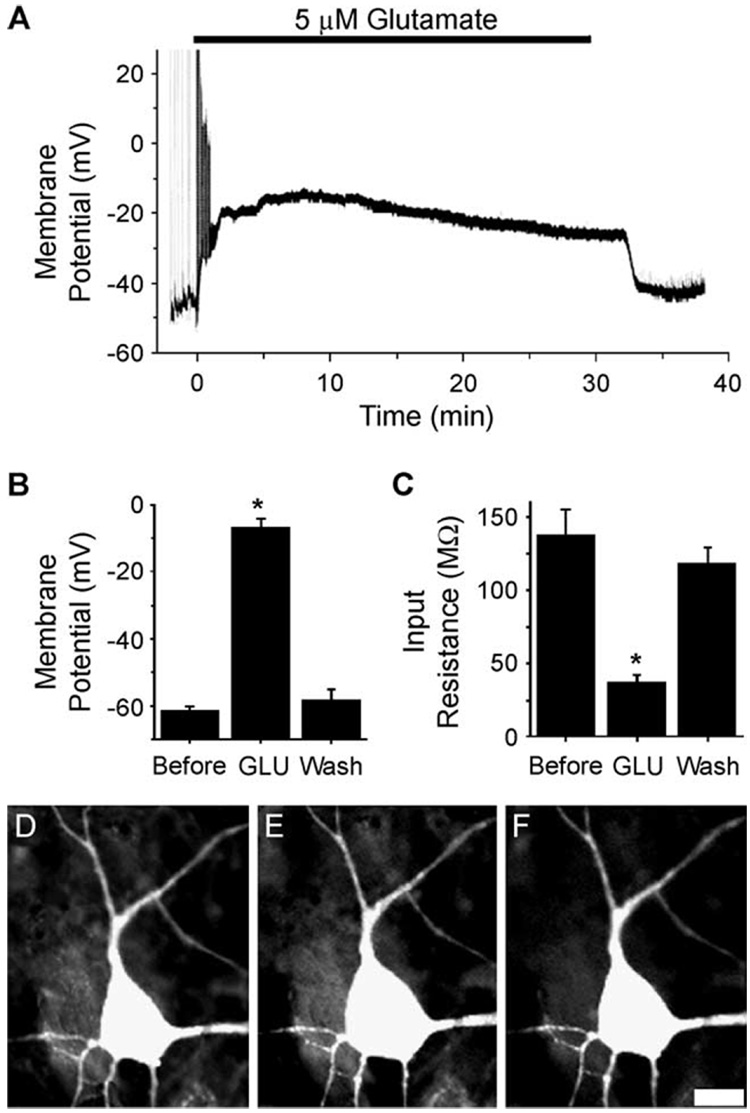
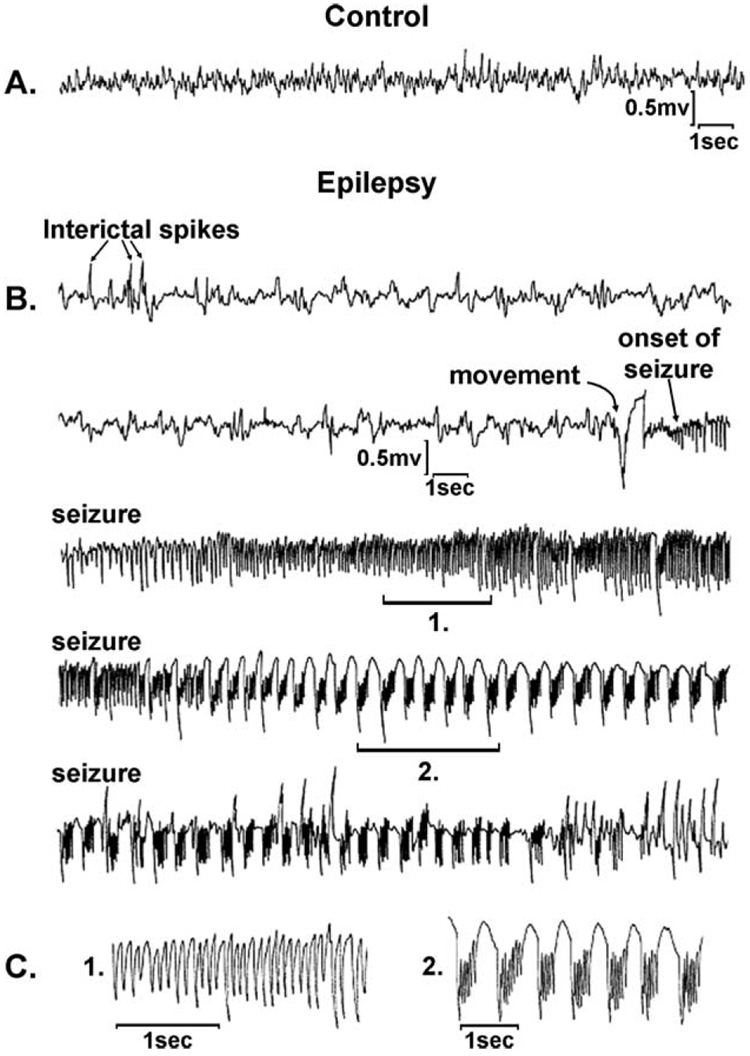
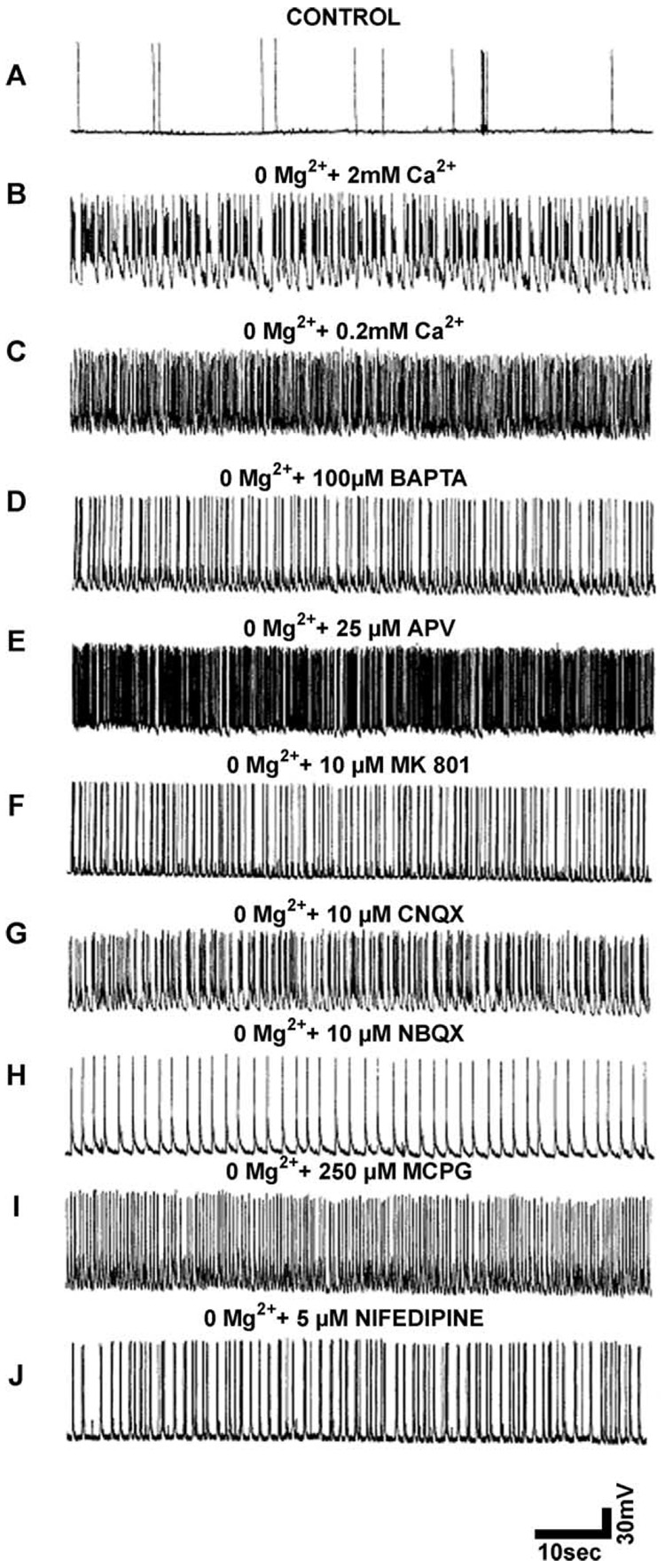
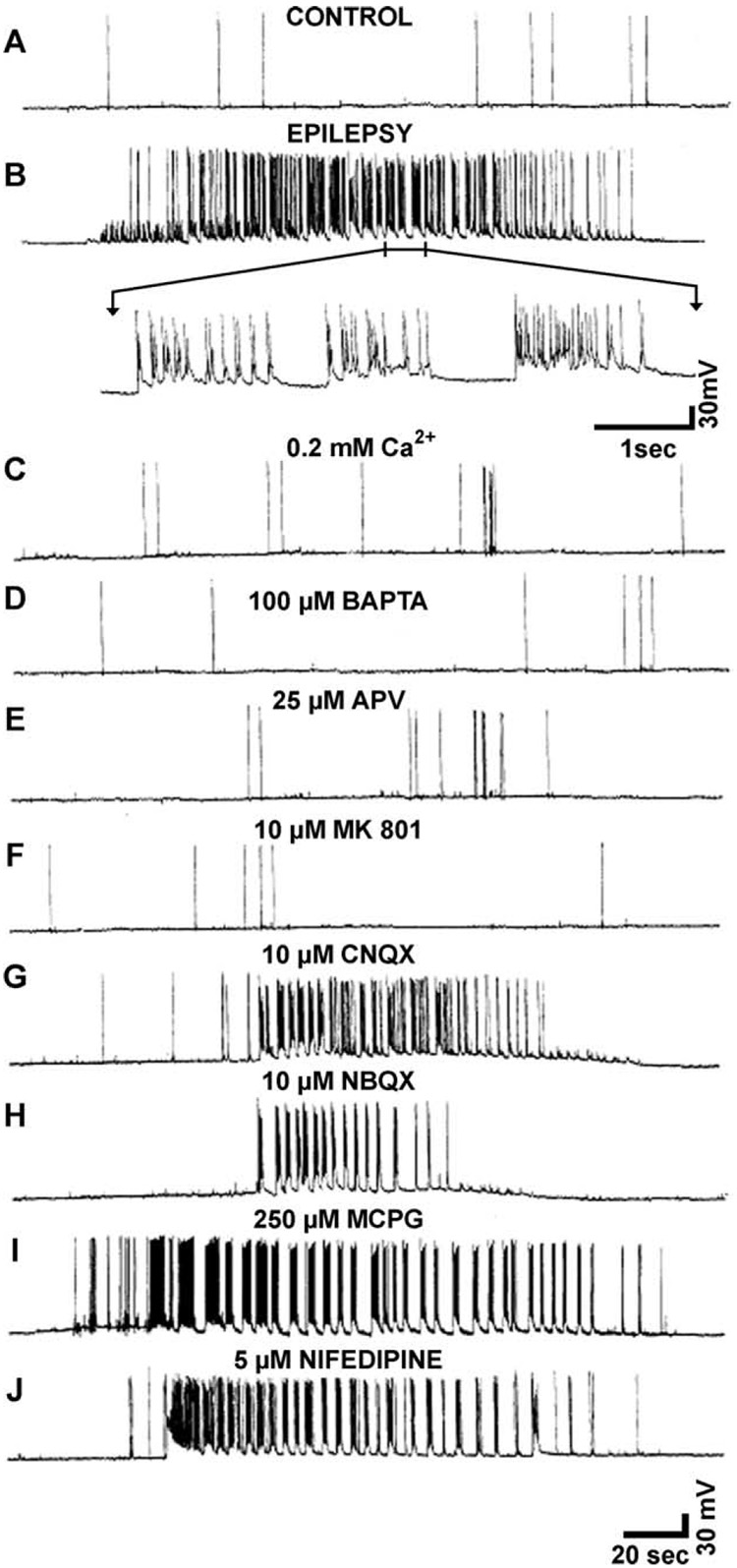
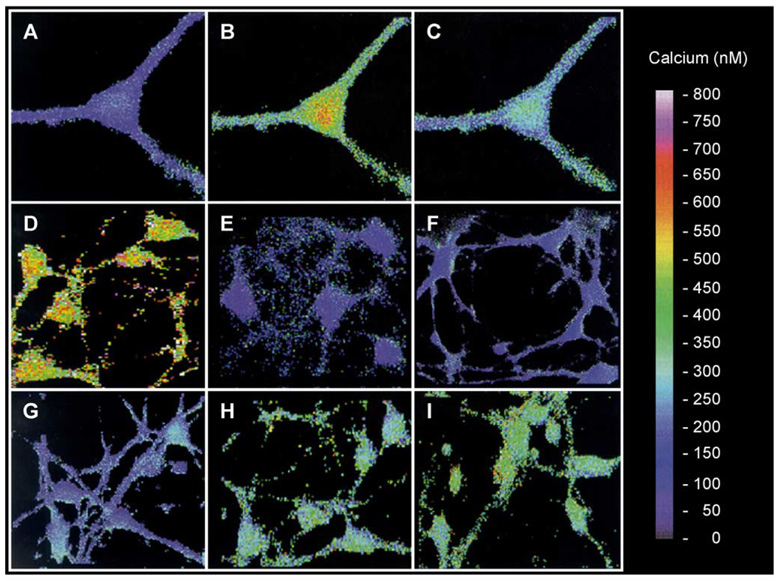
References
-
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115(7):863–877. - PubMed
-
- Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;11(2):180–187. - PubMed
-
- Adams ME, Olivera BM. Neurotoxins: overview of an emerging research technology. Trends Neurosci. 1994;17(4):151–155. - PubMed
-
- Akasu T, Muraoka N, Hasuo H. Hyperexcitability of hippocampal CA1 neurons after fluid percussion injury of the rat cerebral cortex. Neurosci Lett. 2002;329(3):305–308. - PubMed
-
- Albensi BC. Models of brain injury and alterations in synaptic plasticity. J Neurosci Res. 2001;65(4):279–283. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
