

Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation
- PMID: 15738401
- PMCID: PMC553303
- DOI: 10.1073/pnas.0408277102
Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation
Abstract
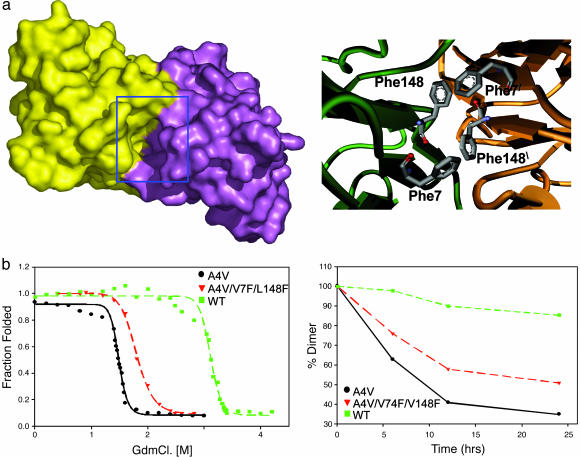
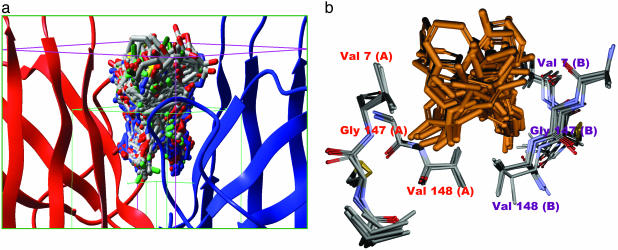
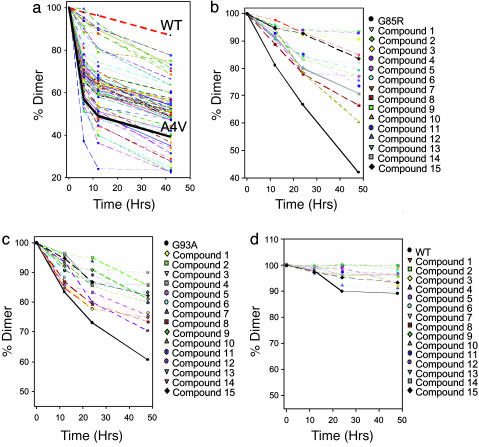
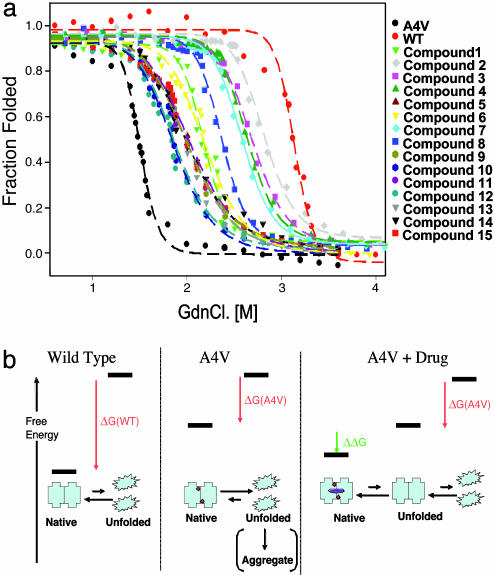
Familial amyotrophic lateral sclerosis (FALS) is a fatal motor neuron disease that is caused by mutations in the gene encoding superoxide dismutase-type 1 (SOD1). The affected regions of the FALS brain are characterized by aggregated SOD1, and the mutations that destabilize SOD1 appear to promote its aggregation in vitro. Because dissociation of the native SOD1 dimer is required for its in vitro aggregation, we initiated an in silico screening program to find drug-like molecules that would stabilize the SOD1 dimer. A potential binding site for such molecules at the SOD1 dimer interface was identified, and its importance was validated by mutagenesis. About 1.5 million molecules from commercial databases were docked at the dimer interface. Of the 100 molecules with the highest predicted binding affinity, 15 significantly inhibited in vitro aggregation and denaturation of A4V, a FALS-linked variant of SOD1. In the presence of several of these molecules, A4V and other FALS-linked SOD1 mutants such as G93A and G85R behaved similarly to wild-type SOD1, suggesting that these compounds could be leads toward effective therapeutics against FALS.
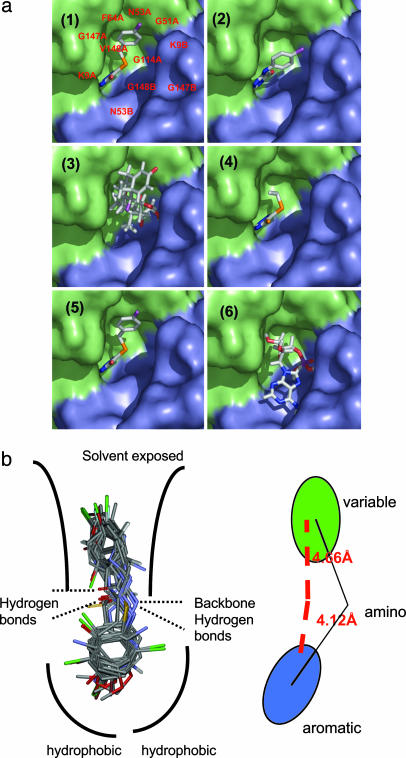
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
