

Structural and mechanistic analysis of two prolyl endopeptidases: role of interdomain dynamics in catalysis and specificity
- PMID: 15738423
- PMCID: PMC553306
- DOI: 10.1073/pnas.0408286102
Structural and mechanistic analysis of two prolyl endopeptidases: role of interdomain dynamics in catalysis and specificity
Abstract
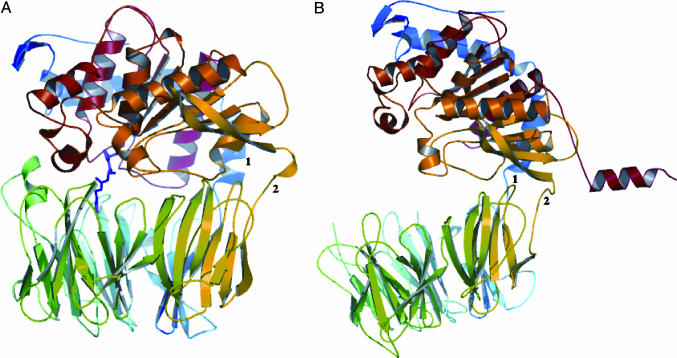
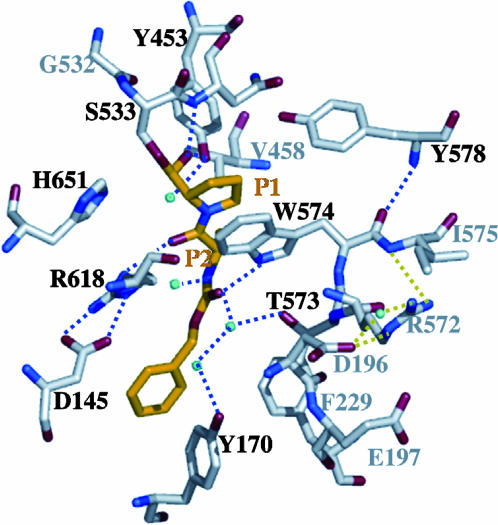
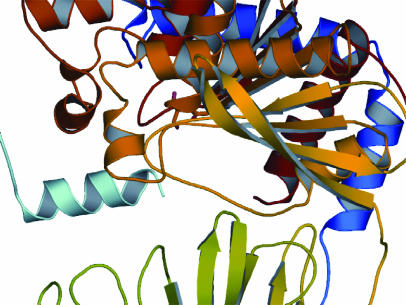
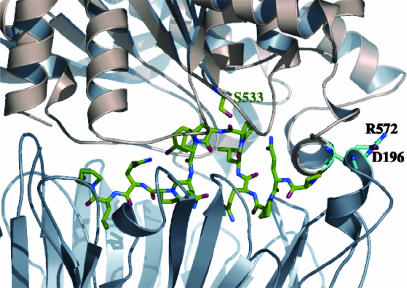
Prolyl endopeptidases (PEPs) are a unique class of serine proteases with considerable therapeutic potential for the treatment of celiac sprue. The crystal structures of two didomain PEPs have been solved in alternative configurations, thereby providing insights into the mode of action of these enzymes. The structure of the Sphingomonas capsulata PEP, solved and refined to 1.8-A resolution, revealed an open configuration of the active site. In contrast, the inhibitor-bound PEP from Myxococcus xanthus was crystallized (1.5-A resolution) in a closed form. Comparative analysis of the two structures highlights a critical role for the domain interface in regulating interdomain dynamics and substrate specificity. Structure-based mutagenesis of the M. xanthus PEP confirms an important role for several interfacial residues. A salt bridge between Arg-572 and Asp-196/Glu-197 appears to act as a latch for opening or closing the didomain enzyme, and Arg-572 and Ile-575 may also help secure the incoming peptide substrate to the open form of the enzyme. Arg-618 and Asp-145 are responsible for anchoring the invariant proline residue in the active site of this postproline-cleaving enzyme. A model is proposed for the docking of a representative substrate PQPQLPYPQPQLP in the active site, where the N-terminal substrate residues interact extensively with the catalytic domain, and the C-terminal residues stretch into the propeller domain. Given the promise of the M. xanthus PEP as an oral therapeutic enzyme for treating celiac sprue, our results provide a strong foundation for further optimization of the PEP's clinically useful features.
Figures
Similar articles
-
Comparative biochemical analysis of three bacterial prolyl endopeptidases: implications for coeliac sprue.Biochem J. 2004 Oct 15;383(Pt 2):311-8. doi: 10.1042/BJ20040907. Biochem J. 2004. PMID: 15245330 Free PMC article.
-
Unraveling the allosteric mechanisms of prolyl endopeptidases for celiac disease therapy: Insights from molecular dynamics simulations.Int J Biol Macromol. 2024 Feb;259(Pt 2):129313. doi: 10.1016/j.ijbiomac.2024.129313. Epub 2024 Jan 10. Int J Biol Macromol. 2024. PMID: 38216012
-
Prolyl endopeptidases.Cell Mol Life Sci. 2007 Feb;64(3):345-55. doi: 10.1007/s00018-006-6317-y. Cell Mol Life Sci. 2007. PMID: 17160352 Free PMC article. Review.
-
Induced-fit mechanism for prolyl endopeptidase.J Biol Chem. 2010 Jul 9;285(28):21487-95. doi: 10.1074/jbc.M109.092692. Epub 2010 May 5. J Biol Chem. 2010. PMID: 20444688 Free PMC article.
-
Prolyl oligopeptidase structure and dynamics.CNS Neurol Disord Drug Targets. 2011 May;10(3):306-10. doi: 10.2174/187152711794653850. CNS Neurol Disord Drug Targets. 2011. PMID: 21222626 Review.
Cited by
-
Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown.Am J Physiol Lung Cell Mol Physiol. 2011 Feb;300(2):L255-65. doi: 10.1152/ajplung.00304.2010. Epub 2010 Nov 26. Am J Physiol Lung Cell Mol Physiol. 2011. PMID: 21112944 Free PMC article.
-
Targeted modification of wheat grain protein to reduce the content of celiac causing epitopes.Funct Integr Genomics. 2012 Aug;12(3):417-38. doi: 10.1007/s10142-012-0287-y. Epub 2012 Jun 26. Funct Integr Genomics. 2012. PMID: 22732824 Review.
-
Oral enzyme therapy for celiac sprue.Methods Enzymol. 2012;502:241-71. doi: 10.1016/B978-0-12-416039-2.00013-6. Methods Enzymol. 2012. PMID: 22208988 Free PMC article.
-
Identification and analysis of multivalent proteolytically resistant peptides from gluten: implications for celiac sprue.J Proteome Res. 2005 Sep-Oct;4(5):1732-41. doi: 10.1021/pr050173t. J Proteome Res. 2005. PMID: 16212427 Free PMC article.
-
The potentials of probiotics on gluten hydrolysis; a review study.Gastroenterol Hepatol Bed Bench. 2020 Winter;13(Suppl1):S1-S7. Gastroenterol Hepatol Bed Bench. 2020. PMID: 33584998 Free PMC article. Review.
References
-
- Yoshimoto, T., Walter, R. & Tsuru, D. (1980) J. Biol. Chem. 255, 4786-4792. - PubMed
-
- Yoshimoto, T., Kanatani, A., Shimoda, T., Inaoka, T., Kokubo, T. & Tsuru, D. (1991) J. Biochem. (Tokyo) 110, 873-878. - PubMed
-
- Yoshimoto, T., Miyazaki, K., Haraguchi, N., Kitazono, A., Kabashima, T. & Ito, K. (1997) Biol. Pharm. Bull. 20, 1047-1050. - PubMed
-
- Kabashima, T., Fujii, M., Meng, Y., Ito, K. & Yoshimoto, T. (1998) Arch. Biochem. Biophys. 358, 141-148. - PubMed
-
- Rennex, D., Hemmings, B. A., Hofsteenge, J. & Stone, S. R. (1991) Biochemistry 30, 2195-2203. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
