

Post-entrapment genome engineering: first exon size does not affect the expression of fusion transcripts generated by gene entrapment
- PMID: 15741512
- PMCID: PMC551569
- DOI: 10.1101/gr.3258105
Post-entrapment genome engineering: first exon size does not affect the expression of fusion transcripts generated by gene entrapment
Abstract
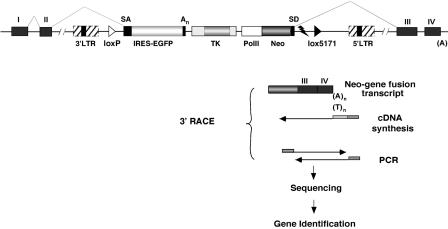
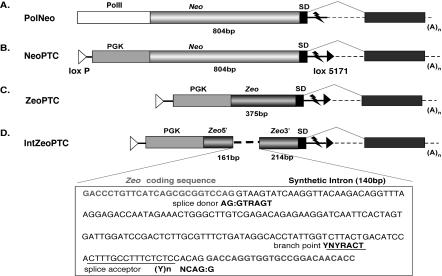
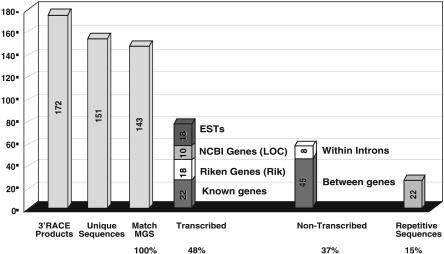
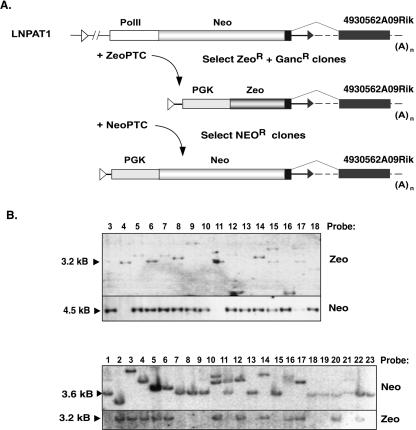
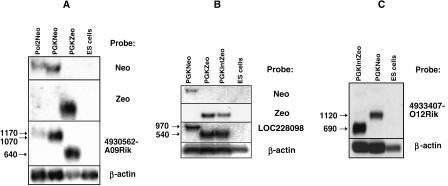
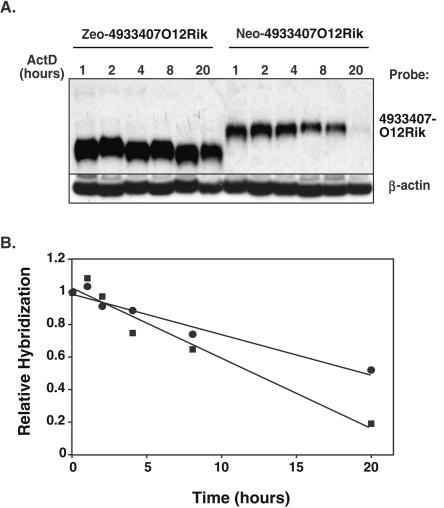
Gene trap mutagenesis in mouse embryonic stem cells has been widely used for genome-wide studies of mammalian gene function. However, while large numbers of genes can be disrupted, individual mutations may suffer from limitations due to the structure and/or placement of targeting vector. To extend the utility of gene trap mutagenesis, replaceable 3' [or poly(A)] gene trap vectors were developed that permit sequences inserted in individual entrapment clones to be engineered by Cre-mediated recombination. 3' traps incorporating different drug resistance genes could be readily exchanged, simply by selecting for the drug-resistance gene of the replacement vector. By substituting different 3' traps, we show that otherwise identical fusion genes containing a large first exon (804 nt) are not expressed at appreciably lower levels than genes expressing small first exons (384 and 151 nt). Thus, size appears to have less effect on the expression and processing of first exons than has been reported for internal exons. Finally, a retroviral poly(A) trap (consisting of a RNA polymerase II promoter, a neomycin-resistance gene, and 5'-splice site) typically produced mutagenized clones in which vector sequences spliced to the 3'-terminal exons of cellular transcription units, suggesting strong selection for fusion transcripts that evade nonsense-mediated decay. The efficient exchange of poly(A) traps should greatly extend the utility of mutant libraries generated by gene entrapment and provides new strategies to study the rules that govern the expression of exons inserted throughout the genome.
Figures
Similar articles
-
Mutagenesis of diploid mammalian genes by gene entrapment.Nucleic Acids Res. 2006;34(20):e139. doi: 10.1093/nar/gkl728. Epub 2006 Oct 24. Nucleic Acids Res. 2006. PMID: 17062627 Free PMC article.
-
Activation of cryptic 3' splice sites within introns of cellular genes following gene entrapment.Nucleic Acids Res. 2004 May 20;32(9):2912-24. doi: 10.1093/nar/gkh604. Print 2004. Nucleic Acids Res. 2004. PMID: 15155860 Free PMC article.
-
Gene trap mutagenesis of hnRNP A2/B1: a cryptic 3' splice site in the neomycin resistance gene allows continued expression of the disrupted cellular gene.BMC Genomics. 2003 Jan 20;4(1):2. doi: 10.1186/1471-2164-4-2. Epub 2003 Jan 20. BMC Genomics. 2003. PMID: 12546712 Free PMC article.
-
The gene trap approach in embryonic stem cells: the potential for genetic screens in mice.Ciba Found Symp. 1992;165:277-88; discussion 288-97. doi: 10.1002/9780470514221.ch16. Ciba Found Symp. 1992. PMID: 1516474 Review.
-
The new mouse genetics: altering the genome by gene targeting.Trends Genet. 1989 Mar;5(3):70-6. doi: 10.1016/0168-9525(89)90029-2. Trends Genet. 1989. PMID: 2660363 Review.
Cited by
-
Expression-independent gene trap vectors for random and targeted mutagenesis in embryonic stem cells.Nucleic Acids Res. 2009 Oct;37(19):e129. doi: 10.1093/nar/gkp640. Epub 2009 Aug 19. Nucleic Acids Res. 2009. PMID: 19692586 Free PMC article.
-
Mutant non-coding RNA resource in mouse embryonic stem cells.Dis Model Mech. 2021 Feb 5;14(2):dmm047803. doi: 10.1242/dmm.047803. Dis Model Mech. 2021. PMID: 33729986 Free PMC article.
-
Mutagenesis of diploid mammalian genes by gene entrapment.Nucleic Acids Res. 2006;34(20):e139. doi: 10.1093/nar/gkl728. Epub 2006 Oct 24. Nucleic Acids Res. 2006. PMID: 17062627 Free PMC article.
-
Comparative analysis of right element mutant lox sites on recombination efficiency in embryonic stem cells.BMC Biotechnol. 2010 Mar 31;10:29. doi: 10.1186/1472-6750-10-29. BMC Biotechnol. 2010. PMID: 20356367 Free PMC article.
-
PiggyBac transposon-based polyadenylation-signal trap for genome-wide mutagenesis in mice.Sci Rep. 2016 Jun 13;6:27788. doi: 10.1038/srep27788. Sci Rep. 2016. PMID: 27292714 Free PMC article.
References
-
- Adra, C.N., Boer, P.H., and McBurney, M.W. 1987. Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene 60: 65-74. - PubMed
-
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. - PubMed
-
- Araki, K., Imaizumi, T., Sekimoto, T., Yoshinobu, K., Yoshimuta, J., Akizuki, M., Miura, K., Araki, M., and Yamamura, K. 1999. Exchangeable gene trap using the Cre/mutated lox system. Cell Mol. Biol. 45: 737-750. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials