

Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors
- PMID: 15753211
- PMCID: PMC2212829
- DOI: 10.1084/jem.20041684
Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors
Abstract
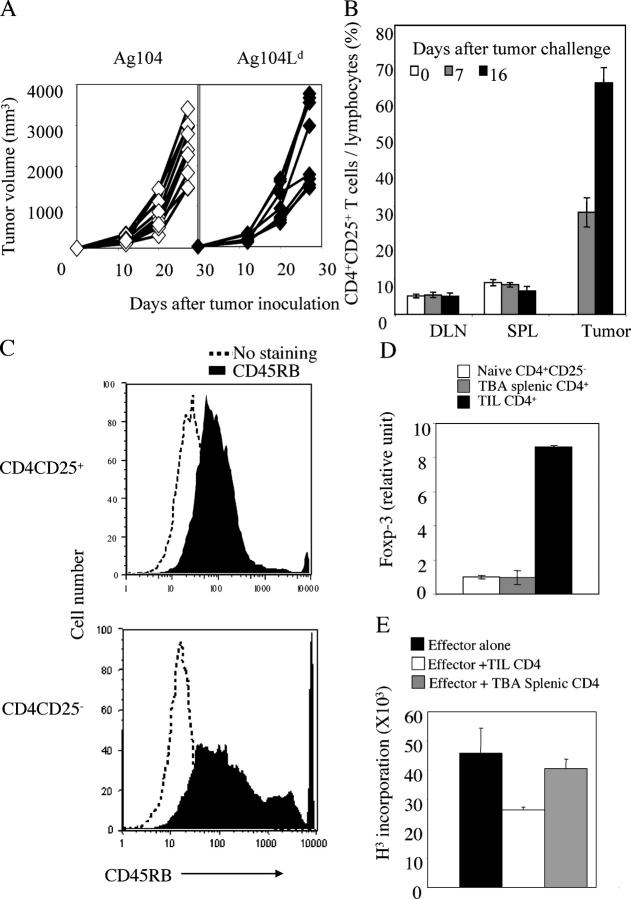
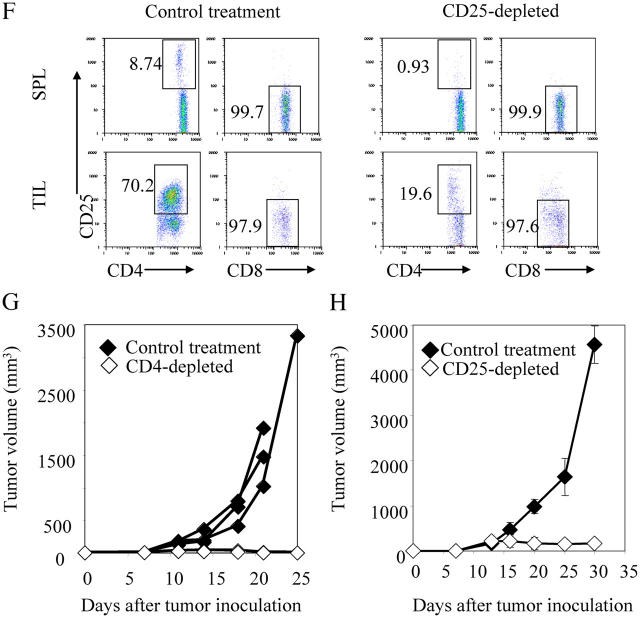
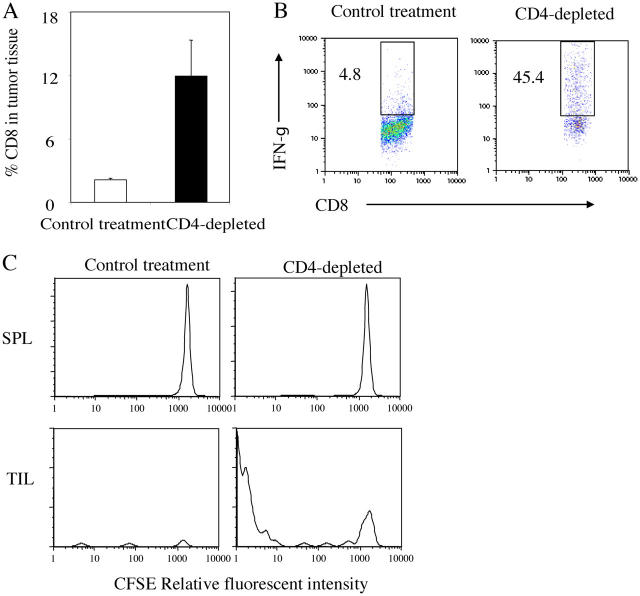
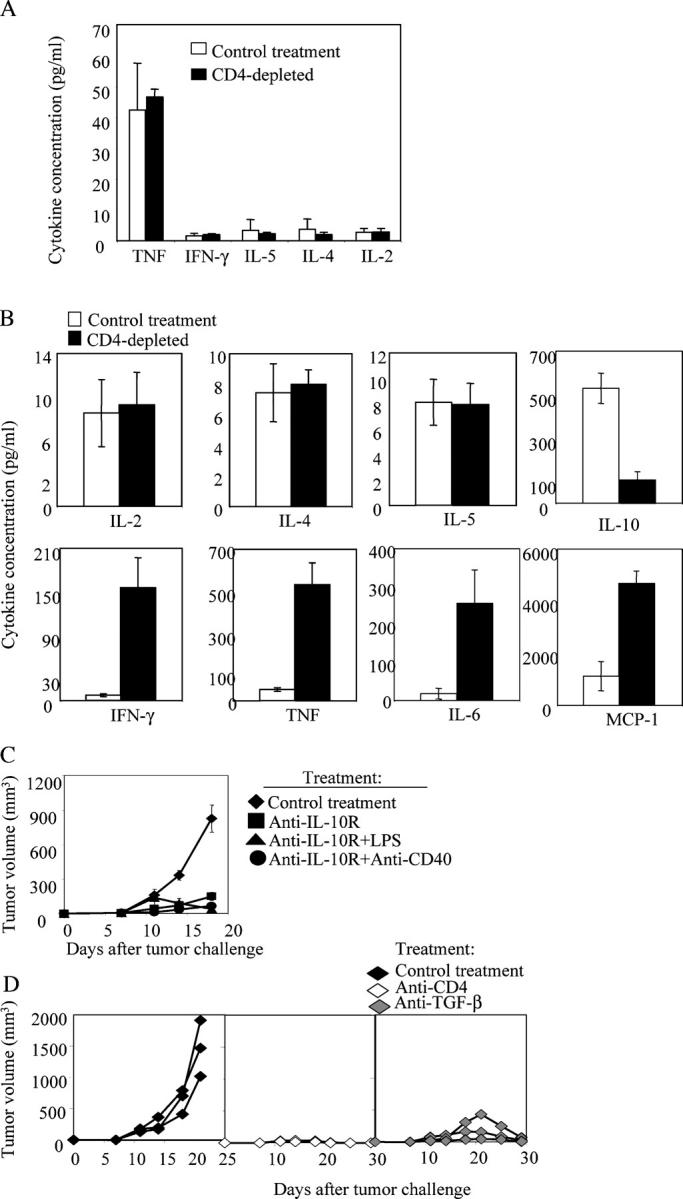
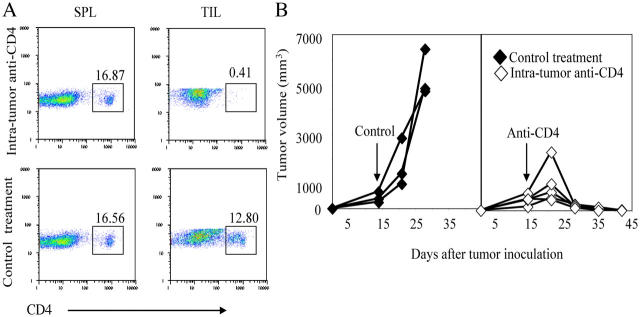
Tumor environment can be critical for preventing the immunological destruction of antigenic tumors. We have observed a selective accumulation of CD4(+)CD25(+) T cells inside tumors. In a murine fibrosarcoma L(d)-expressing Ag104, these cells made up the majority of tumor-infiltrating lymphocytes at the late stage of tumor progression, and their depletion during the effector phase, rather than priming phase, successfully enhanced antitumor immunity. We show here that CD4(+)CD25(+) T cells suppressed the proliferation and interferon-gamma production of CD8(+) T cells in vivo at the local tumor site. Blockade of the effects of IL-10 and TGF-beta partially reversed the suppression imposed by the CD4(+) cells. Furthermore, local depletion of CD4(+) cells inside the tumor resulted in a change of cytokine milieu and led to the eradication of well-established highly aggressive tumors and the development of long-term antitumor memory. Therefore, CD4(+)CD25(+) T cells maintained an environment in the tumor that concealed the immunogenicity of tumor cells to permit progressive growth of antigenic tumors. Our study illustrates that the suppression of antitumor immunity by regulatory T cells occurs predominantly at the tumor site, and that local reversal of suppression, even at a late stage of tumor development, can be an effective treatment for well-established cancers.
Figures
References
-
- Dunn, G.P., L.J. Old, and R.D. Schreiber. 2004. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22:329–360. - PubMed
-
- Rosenberg, S.A. 1999. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 10:281–287. - PubMed
-
- Pardoll, D. 2003. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 21:807–839. - PubMed
-
- Monach, P.A., S.C. Meredith, C.T. Siegel, and H. Schreiber. 1995. A unique tumor antigen produced by a single amino acid substitution. Immunity. 2:45–59. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
