

Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors
- PMID: 15758166
- PMCID: PMC6725188
- DOI: 10.1523/JNEUROSCI.4998-04.2005
Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors
Abstract
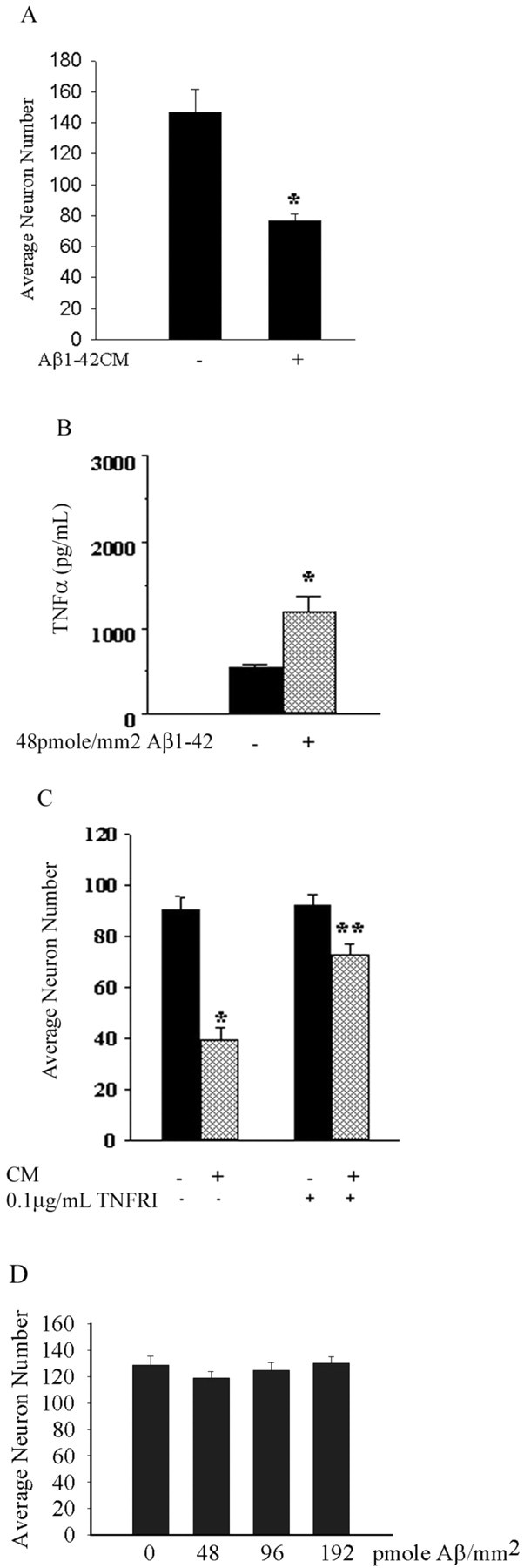
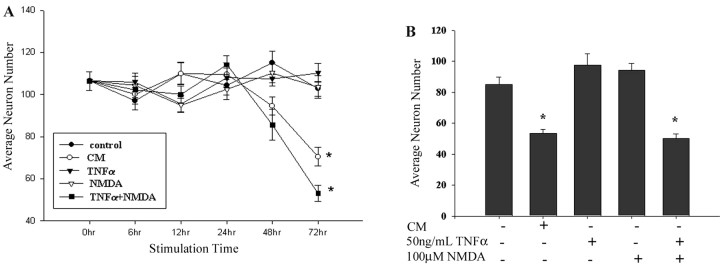
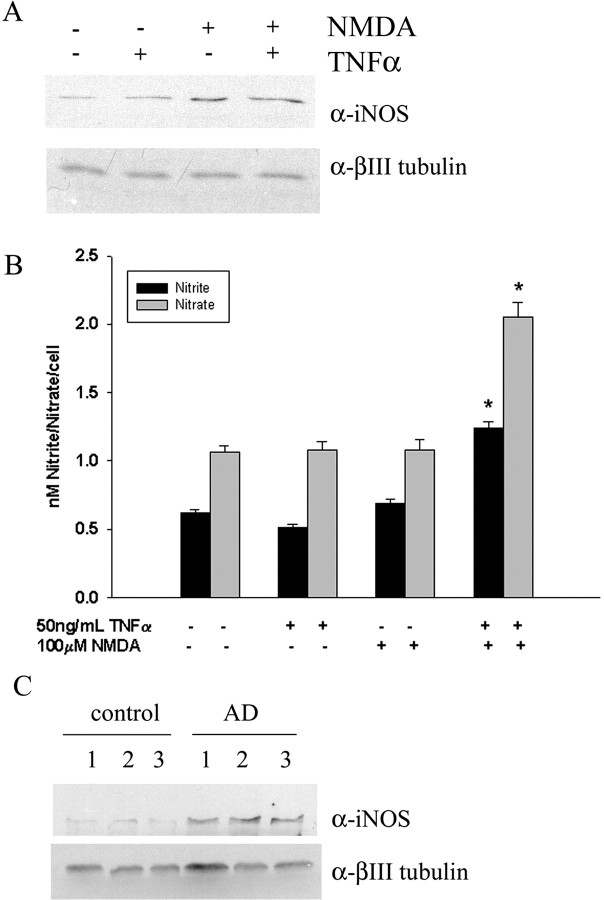
Although abundant reactive microglia are found associated with beta-amyloid (Abeta) plaques in Alzheimer's disease (AD) brains, their contribution to cell loss remains speculative. A variety of studies have documented the ability of Abeta fibrils to directly stimulate microglia in vitro to assume a neurotoxic phenotype characterized by secretion of a plethora of proinflammatory molecules. Collectively, these data suggest that activated microglia play a direct role in contributing to neuron death in AD rather than simply a role in clearance after plaque deposition. Although it is clear the Abeta-stimulated microglia acutely secrete toxic oxidizing species, the identity of longer-lived neurotoxic agents remains less defined. We used Abeta-stimulated conditioned media from primary mouse microglia to identify more stable neurotoxic secretions. The NMDA receptor antagonists memantine and 2-amino-5-phosphopetanoic acid as well as soluble tumor necrosis factor alpha (TNFalpha) receptor protect neurons from microglial-conditioned media-dependent death, implicating the excitatory neurotransmitter glutamate and the proinflammatory cytokine TNFalpha as effectors of microglial-stimulated death. Neuron death occurs in an oxidative damage-dependent manner, requiring activity of inducible nitric oxide synthase. Toxicity results from coincident stimulation of the TNFalpha and NMDA receptors, because stimulations of either alone are insufficient to initiate cell death. These findings suggest the hypothesis that AD brains provide the appropriate microglial-mediated inflammatory environment for TNFalpha and glutamate to synergistically stimulate toxic activation of their respective signaling pathways in neurons as a contributing mechanism of cell death.
Figures






References
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. - PMC - PubMed
-
- Barger S, Basile AS (2001) Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem 76: 846-854. - PubMed
-
- Barger SW, Hortser D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP (1995) Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA 92: 9328-9332. - PMC - PubMed
-
- Beckman J, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin J, Tsai M (1992) Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys 298: 438-445. - PubMed
-
- Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F (1999) Beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem 274: 15493-15499. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources