

Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones
- PMID: 15760935
- PMCID: PMC1464474
- DOI: 10.1113/jphysiol.2005.084780
Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones
Abstract
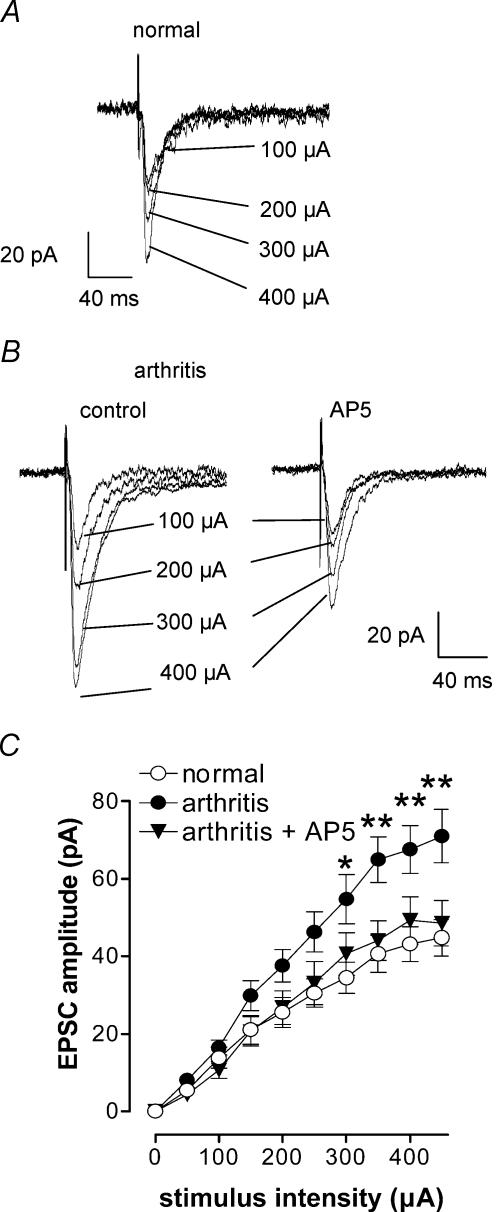
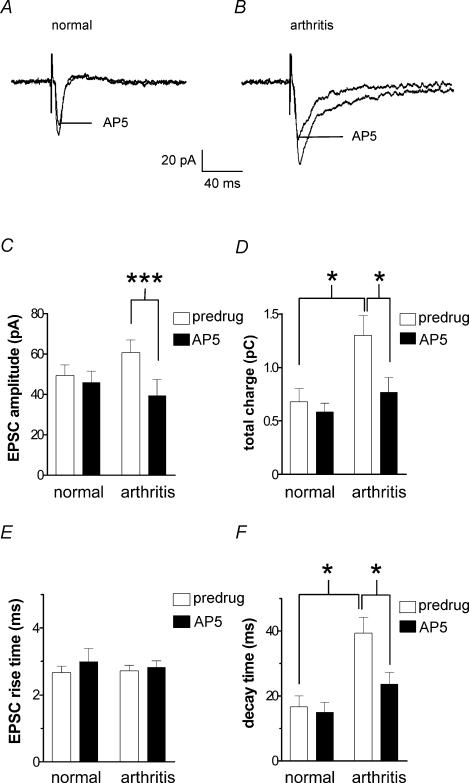
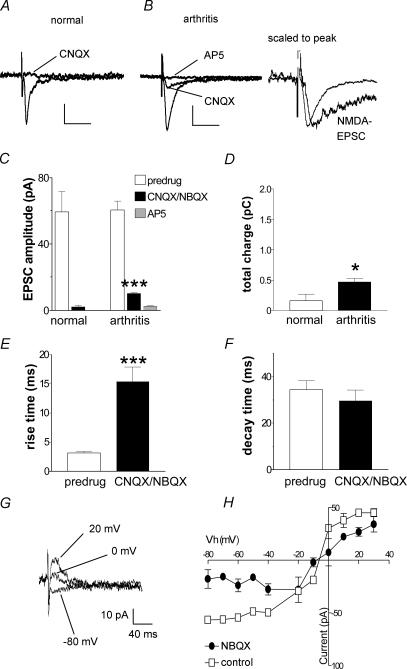
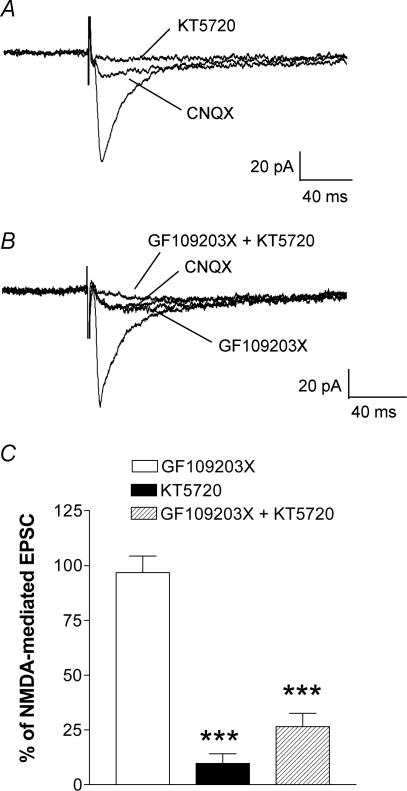
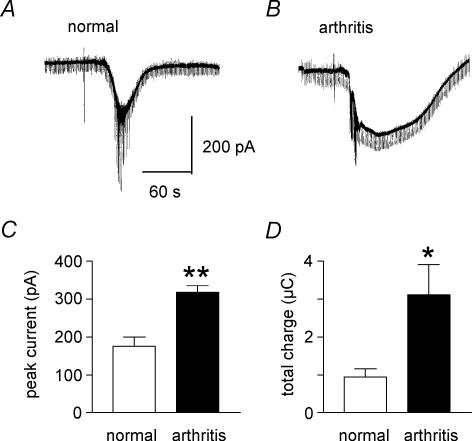
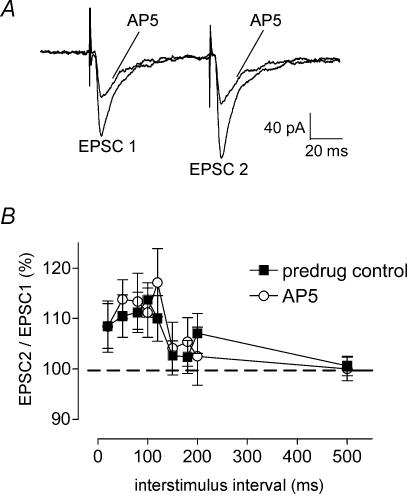
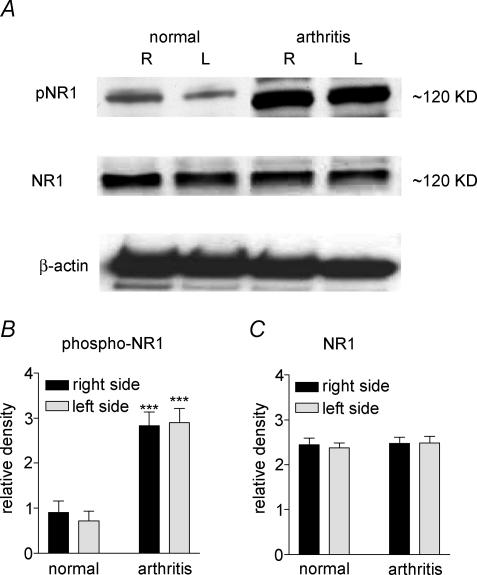
Mechanisms of pain-related plasticity in the amygdala, a key player in emotionality, were studied at the cellular and molecular levels in a model of arthritic pain. The influence of the arthritis pain state induced in vivo on synaptic transmission and N-methyl-d-aspartate (NMDA) receptor function was examined in vitro using whole-cell voltage-clamp recordings of neurones in the latero-capsular part of the central nucleus of the amygdala (CeA), which is now defined as the 'nociceptive amygdala'. Synaptic transmission was evoked by electrical stimulation of afferents from the pontine parabrachial area (part of the spino-parabrachio-amygdaloid pain pathway) in brain slices from control rats and from arthritic rats. This study shows that pain-related synaptic plasticity is accompanied by protein kinase A (PKA)-mediated enhanced NMDA-receptor function and increased phosphorylation of NMDA-receptor 1 (NR1) subunits. Synaptic plasticity in the arthritis pain model, but not normal synaptic transmission in control neurones, was inhibited by a selective NMDA receptor antagonist. Accordingly, an NMDA receptor-mediated synaptic component was recorded in neurones from arthritic animals, but not in control neurones, and was blocked by inhibition of PKA but not protein kinase C (PKC). Exogenous NMDA evoked a larger inward current in neurones from arthritic animals than in control neurones, indicating a postsynaptic effect. Paired-pulse facilitation, a measure of presynaptic mechanisms, was not affected by an NMDA-receptor antagonist. Increased levels of phosphorylated NR1 protein, but not of total NR1, were measured in the CeA of arthritic rats compared to controls. Our results suggest that pain-related synaptic plasticity in the amygdala involves a critical switch of postsynaptic NMDA receptor function through PKA-dependent NR1 phosphorylation.
Figures
References
-
- Becerra LR, Breiter HC, Stojanovic M, Fishman S, Edwards A, Comite AR, Gonzalez RG. Human brain activation under controlled thermal stimulation and habituation of noxious heat: an fMRI study. Magn Reson Med. 1999;41:1044–1057. - PubMed
-
- Bingel U, Quante M, Knab R, Bromm B, Weiller C, Buchel C. Subcortical structures involved in pain processing: evidence from single-trial fMRI. Pain. 2002;99:313–321. - PubMed
-
- Bornhovd K, Quante M, Glauche V, Bromm B, Weiller C, Buchel C. Painful stimuli evoke different stimulus-response functions in the amygdala, prefrontal, insula and somatosensory cortex: a single-trial fMRI study. Brain. 2002;125:1326–1336. - PubMed
-
- Bourgeais L, Gauriau C, Bernard J-F. Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci. 2001;14:229–255. - PubMed
-
- Burnashev N, Schoepfer R, Monyer H, Ruppersberg JP, Gunther W, Seeburg PH, Sakmann B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257:1415–1419. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources