

Review
doi: 10.1172/JCI24459.
NO/redox disequilibrium in the failing heart and cardiovascular system
Affiliations
- PMID: 15765132
- PMCID: PMC1052013
- DOI: 10.1172/JCI24459
Item in Clipboard
Review
NO/redox disequilibrium in the failing heart and cardiovascular system
J Clin Invest.
2005 Mar.
Abstract
There is growing evidence that the altered production and/or spatiotemporal distribution of reactive oxygen and nitrogen species creates oxidative and/or nitrosative stresses in the failing heart and vascular tree, which contribute to the abnormal cardiac and vascular phenotypes that characterize the failing cardiovascular system. These derangements at the integrated system level can be interpreted at the cellular and molecular levels in terms of adverse effects on signaling elements in the heart, vasculature, and blood that subserve cardiac and vascular homeostasis.
Figures
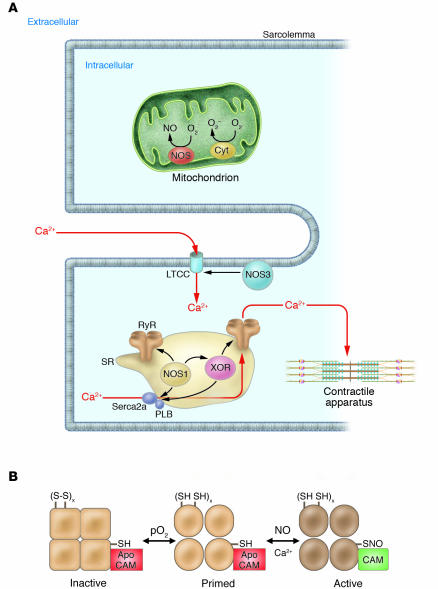
(A) Spatial localization of NOSs and oxidases in the cardiac myocyte. NOS3 localizes to the sarcolemmal caveolae (85, 97), where it participates in regulation of L-type Ca2+ channel (LTCC) currents, mediated either by cGMP formation (S64) or by S-nitrosylation of the LTCC (S13). NOS1 localizes to the SR (32), where it facilitates the SR Ca2+ cycle (97) by S-nitrosylation of the RyR and possibly the SR Ca2+ ATPase (SERCA2a) (17, 33). XOR also localizes to the SR in the cardiac myocyte; upregulation of protein or activity (caused by SR NOS1 deficiency) disrupts SNO regulation of the RyR (22). Other oxidases (e.g., NADPH oxidase) have been described in the cardiac myocyte (50), but precise signaling roles, identities, and/or subcellular localizations have not been elucidated. The mitochondria are an additional source of both O2– and NO, which may participate in control of mitochondrial respiration (S65–S67) and apoptosis (S41). Cyt, cytochrome; PLB, phospholamban. (B) Regulation of the RyR by S-nitrosylation. S-nitrosylation occurs at a single cysteine residue (1 of approximately 50 free thiols), which resides within a calmodulin-binding domain of the cardiac RyR (17). NO binding (shown for RyR1 of skeletal muscle) occurs in an oxygen-concentration–dependent manner and primes the channel for calmodulin regulation (18). Higher pO2 oxidizes a small set of RyR-associated thiols that regulate the channel’s responsiveness to NO. SH, reduced thiol; S-S, oxidized thiols; CAM, calmodulin; x refers to a small set between 1 and 3.
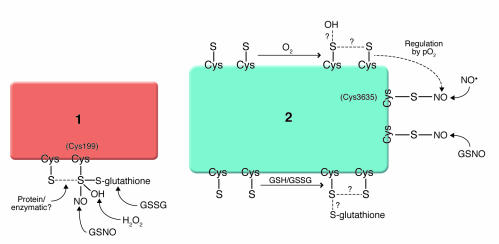
NO/redox-based signaling and nitrosative stress. Molecular recognition by cysteine-containing proteins is achieved either through the existence of single classes of thiols that are adapted to differentiate NO modification (S-nitrosylation) from oxidations (S-glutathionylation, S-S [intramolecular disulfide] and/or sulfur oxides [SOx–, where x is 1–3]) – exemplified in protein 1 – or through the presence of multiple classes of thiols, each adapted to recognize different redox-related molecules, including NO, GSNO, H2O2, O2, and cellular redox potential (for protein 2, note that some classes of thiols may be functionally linked to others, exemplified in the pO2-dependent oxidation of RyR thiols that promotes S-nitrosylation). In model 1, thiol oxidation would adversely impact nitrosylation signaling. In model 2, signal malfunction may result from altered amounts, timing, and/or the nature of RNS/ROS-based modifications. S, cysteine thiol; GSH/GSSG, glutathione/glutathione disulfide; pO2, partial pressure of O2.
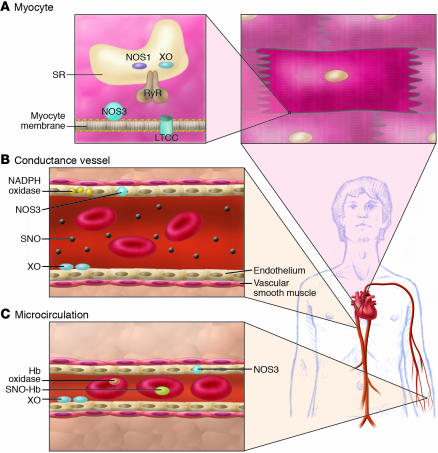
Consequences of NO/redox disequilibrium in the cardiovascular system – congestive HF phenotype. The balance between nitric oxide (NOS and hemoglobin–based activities) and superoxide/ROS production (oxidase activity) plays a pivotal role in cell/organ function at key sites in the cardiovascular system, including the heart (A), large- and medium-sized conductance blood vessels (B), and the microvasculature (C) (S12). At each of these sites, NO/redox disequilibrium is identified with dysregulated NO-based signaling. (A) In the cardiac myocyte, NO regulates receptor-mediated signal transduction, the calcium cycle, mitrochondrial respiration, and myofilament contractility. Loss of NOS in the cardiac SR (95) impairs NO signaling and creates oxidative stress (by relieving inhibition of the oxidase) (22). Upregulation of inducible NOS (NOS2) may further disrupt physiologic NO regulation by producing a nitrosative stress. The NO/redox disequilibrium that ensues in HF is characterized by disruption and/or impairment of the cardiac calcium cycle, mitochondrial respiration, and myofilament responsiveness to activator calcium. (B) In conductance vessels, vasoconstriction may result from diminished endothelial NOS activity and/or impaired delivery of plasma-borne NO bioactivity. A NO/redox disequilibrium is linked to increased expression or activity of both vascular NADPH oxidase (Nox4) (99) and circulating XO (46). (C) In the microvasculature, rbcs govern NO bioactivity. Lower venous O2 saturation in HF may subserve a NO/redox disequilibrium by impairing NO release from rbcs (SNO-Hb) and promoting hemoglobin oxidase activity (44, 75). Impaired vasodilation by rbcs may exacerbate tissue ischemia.
Similar articles
-
Nitric oxide in blood. The nitrosative-oxidative disequilibrium hypothesis on the pathogenesis of cardiovascular disease.FEBS J. 2007 Feb;274(4):906-23. doi: 10.1111/j.1742-4658.2007.05660.x. Epub 2007 Jan 22. FEBS J. 2007. PMID: 17244198 Review.
-
Nitric oxide and cell signaling: modulation of redox tone and protein modification.Amino Acids. 2003 Dec;25(3-4):313-21. doi: 10.1007/s00726-003-0019-7. Epub 2003 Aug 28. Amino Acids. 2003. PMID: 14661093 Review.
-
Redox-mediated signal transduction by cardiovascular Nox NADPH oxidases.J Mol Cell Cardiol. 2014 Aug;73:70-9. doi: 10.1016/j.yjmcc.2014.02.006. Epub 2014 Feb 19. J Mol Cell Cardiol. 2014. PMID: 24560815 Review.
-
Redox regulation of cardiovascular inflammation - Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species.Free Radic Biol Med. 2017 Aug;109:48-60. doi: 10.1016/j.freeradbiomed.2017.01.027. Epub 2017 Jan 18. Free Radic Biol Med. 2017. PMID: 28108279 Review.
-
Oxygen, oxidative stress, hypoxia, and heart failure.J Clin Invest. 2005 Mar;115(3):500-8. doi: 10.1172/JCI24408. J Clin Invest. 2005. PMID: 15765131 Free PMC article. Review.
Cited by
-
Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery.Chem Rev. 2013 Jul 10;113(7):4633-79. doi: 10.1021/cr300163e. Epub 2013 Mar 20. Chem Rev. 2013. PMID: 23514336 Free PMC article. Review. No abstract available.
-
Implications of Oxidative and Nitrosative Post-Translational Modifications in Therapeutic Strategies against Reperfusion Damage.Antioxidants (Basel). 2021 May 8;10(5):749. doi: 10.3390/antiox10050749. Antioxidants (Basel). 2021. PMID: 34066806 Free PMC article. Review.
-
Alterations of tumor microenvironment by nitric oxide impedes castration-resistant prostate cancer growth.Proc Natl Acad Sci U S A. 2018 Oct 30;115(44):11298-11303. doi: 10.1073/pnas.1812704115. Epub 2018 Oct 15. Proc Natl Acad Sci U S A. 2018. PMID: 30322928 Free PMC article.
-
Deletion of metallothionein exacerbates intermittent hypoxia-induced oxidative and inflammatory injury in aorta.Oxid Med Cell Longev. 2014;2014:141053. doi: 10.1155/2014/141053. Epub 2014 Aug 6. Oxid Med Cell Longev. 2014. PMID: 25177426 Free PMC article.
-
Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential.Cells. 2025 Feb 20;14(5):324. doi: 10.3390/cells14050324. Cells. 2025. PMID: 40072053 Free PMC article. Review.
References
-
- McCord JM, Fridovitch I. The reduction of cytochrome c by milk xanthine oxidase. J. Biol. Chem. 1968;243:5753–5760. - PubMed
-
- McCord JM, Fridovitch I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J. Biol. Chem. 1969;244:6049–6055. - PubMed
-
- Keith M, et al. Increased oxidative stress in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998;31:1352–1356. - PubMed
-
- Cesselli D, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 2001;89:279–286. - PubMed
-
- Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J. Am. Coll. Cardiol. 1996;28:506–514. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
