

Review
doi: 10.1172/JCI24351.
Genetic causes of human heart failure
Affiliations
- PMID: 15765133
- PMCID: PMC1052010
- DOI: 10.1172/JCI24351
Item in Clipboard
Review
Genetic causes of human heart failure
J Clin Invest.
2005 Mar.
Abstract
Factors that render patients with cardiovascular disease at high risk for heart failure remain incompletely defined. Recent insights into molecular genetic causes of myocardial diseases have highlighted the importance of single-gene defects in the pathogenesis of heart failure. Through analyses of the mechanisms by which a mutation selectively perturbs one component of cardiac physiology and triggers cell and molecular responses, studies of human gene mutations provide a window into the complex processes of cardiac remodeling and heart failure. Knowledge gleaned from these studies shows promise for defining novel therapeutic targets for genetic and acquired causes of heart failure.
Figures
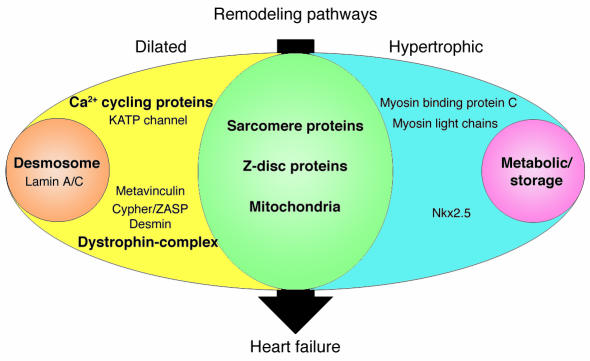
Human gene mutations can cause cardiac hypertrophy (blue), dilation (yellow), or both (green). In addition to these two patterns of remodeling, particular gene defects produce hypertrophic remodeling with glycogen accumulation (pink) or dilated remodeling with fibrofatty degeneration of the myocardium (orange). Sarcomere proteins denote β-myosin heavy chain, cardiac troponin T, cardiac troponin I, α-tropomyosin, cardiac actin, and titin. Metabolic/storage proteins denote AMP-activated protein kinase γ subunit, LAMP2, lysosomal acid α 1,4–glucosidase, and lysosomal hydrolase α-galactosidase A. Z-disc proteins denote MLP and telethonin. Dystrophin-complex proteins denote δ-sarcoglycan, β-sarcoglycan, and dystrophin. Ca2+ cycling proteins denote PLN and RyR2. Desmosome proteins denote plakoglobin, desmoplakin, and plakophilin-2.
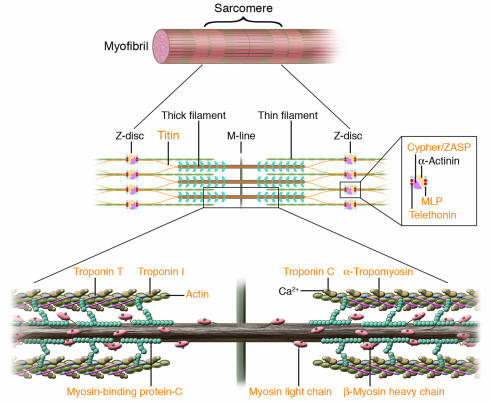
Human mutations affecting contractile and Z-disc proteins. The schematic depicts one sarcomere, the fundamental unit of contraction encompassing the protein segment between flanking Z discs. Sarcomere thin filament proteins are composed of actin and troponins C, T, and I. Sarcomere thick filament proteins include myosin heavy chain, myosin essential and regulatory light chains, myosin-binding protein-C and titin. The sarcomere is anchored through titin and actin interactions with Z disc proteins α-actinin, calsarcin-1, MLP, telethonin (T-cap), and ZASP. Human mutations (orange text) in contractile proteins and Z-disc proteins can cause HCM or DCM.
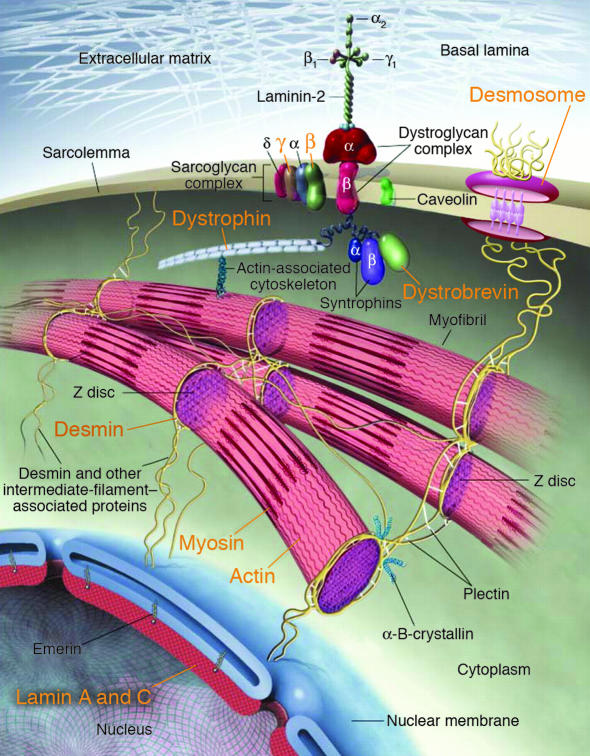
Human mutations (orange text) in components of myocyte cytoarchitecture cause DCM and heart failure. Force produced by sarcomeric actin-myosin interactions is propagated through the actin cytoskeleton and dystrophin to the dystrophin-associated glycoprotein complex (composed of α- and β-dystroglycans, α-, β-, γ- and δ-sarcoglycans, caveolin-3, syntrophin, and dystrobrevin). Desmosome proteins plakoglobin, desmoplakin, and plakophilin-2, provide functional and structural contacts between adjacent cells and are linked through intermediate filament proteins, including desmin, to the nuclear membrane, where lamin A/C is localized. Adapted from ref. .
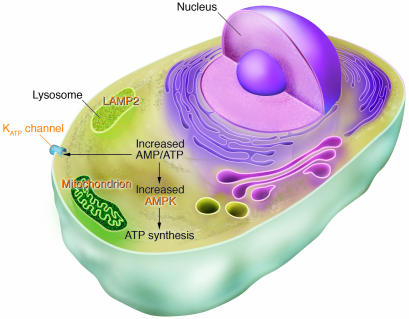
Human gene mutations affecting cardiac energetics and metabolism. Energy substrate utilization is directed by critical metabolic sensors in myocytes, including AMP-activated protein kinase (AMPK), which, in response to increased AMP/ATP levels, phosphorylates target proteins and thereby regulates glycogen and fatty acid metabolism, critical energy sources for the heart. Glycogen metabolism involves a large number of proteins including α-galactosidase A (mutated in Fabry disease) and LAMP2 (mutated in Danon disease). Glycogen and fatty acids are substrates for multiprotein complexes located within the mitochondria for the synthesis of ATP. KATP channels composed of an enzyme complex and a potassium pore participate in decoding metabolic signals to maximize cellular functions during stress adaptation. Human mutations (orange text) that cause cardiomyopathies have been identified in the regulatory SUR2A subunit of KATP, the γ2 subunit of AMPK, mitochondrial proteins, α-galactosidase A, and LAMP2.
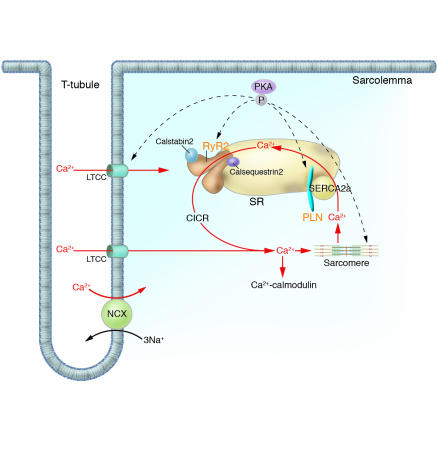
Human mutations affecting Ca2+ cycling proteins. Intracellular Ca2+ handling is the central coordinator of cardiac contraction and relaxation. Ca2+ entering through L-type channels (LTCC) triggers Ca2+ release (CICR) from the SR via the RyR2, and sarcomere contraction is initiated. Relaxation occurs with SR Ca2+ reuptake through the SERCA2a. Calstabin2 coordinates excitation and contraction by modulating RyR2 release of Ca2+. PLN, an SR transmembrane inhibitor of SERCA2a modulates Ca2+ reuptake. Dynamic regulation of these molecules is effected by PKA-mediated phosphorylation. Ca2+ may further function as a universal signaling molecule, stimulating Ca2+-calmodulin and other molecular cascades. Human mutations (orange text) in molecules involved in calcium cycling cause cardiac remodeling and heart failure. NCX, sodium/calcium exchanger.
References
-
- American Heart Association. 2004. Heart disease and stroke statistics – 2004 update. American Heart Association. http://www.americanheart.org.
-
- Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104:557–567. - PubMed
-
- CardioGenomics. Genomics of cardiovascular development, adaptation, and remodeling. NHLBI program for genomic applications. Harvard Medical School. http://cardiogenomics.med.harvard.edu.
-
- Morita H, et al. Molecular epidemiology of hypertrophic cardiomyopathy. Cold Spring Harb. Symp. Quant. Biol. 2002;67:383–388. - PubMed
-
- Richard P, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
