

Protein kinase cascades in the regulation of cardiac hypertrophy
- PMID: 15765134
- PMCID: PMC1052008
- DOI: 10.1172/JCI24178
Protein kinase cascades in the regulation of cardiac hypertrophy
Abstract
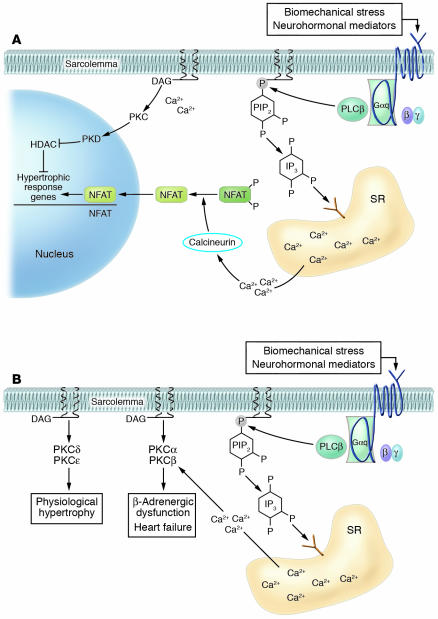
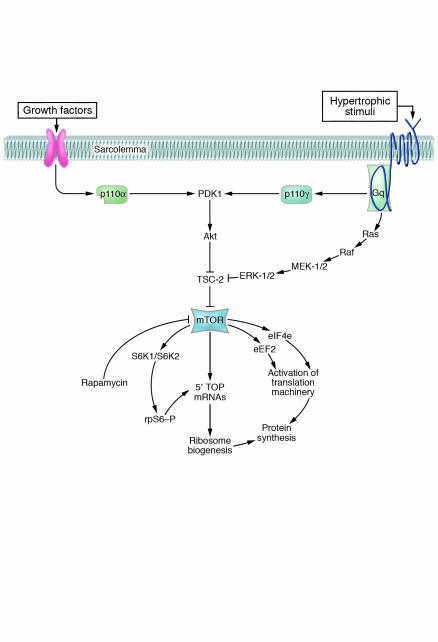
In broad terms, there are 3 types of cardiac hypertrophy: normal growth, growth induced by physical conditioning (i.e., physiologic hypertrophy), and growth induced by pathologic stimuli. Recent evidence suggests that normal and exercise-induced cardiac growth are regulated in large part by the growth hormone/IGF axis via signaling through the PI3K/Akt pathway. In contrast, pathological or reactive cardiac growth is triggered by autocrine and paracrine neurohormonal factors released during biomechanical stress that signal through the Gq/phospholipase C pathway, leading to an increase in cytosolic calcium and activation of PKC. Here we review recent developments in the area of these cardiotrophic kinases, highlighting the utility of animal models that are helping to identify molecular targets in the human condition.
Figures


References
-
- Sadoshima J, Izumo S. Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993;73:413–423. - PubMed
-
- Shubeita HE, et al. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy. J. Biol. Chem. 1990;265:20555–20562. - PubMed
-
- Bristow MR. beta-adrenergic receptor blockade in chronic heart failure. Circulation. 2000;101:558–569. - PubMed
-
- Mehra MR, Uber PA, Francis GS. Heart failure therapy at a crossroad: are there limits to the neurohormonal model? J. Am. Coll. Cardiol. 2003;41:1606–1610. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
