

Toward transcriptional therapies for the failing heart: chemical screens to modulate genes
- PMID: 15765135
- PMCID: PMC1052006
- DOI: 10.1172/JCI24144
Toward transcriptional therapies for the failing heart: chemical screens to modulate genes
Abstract
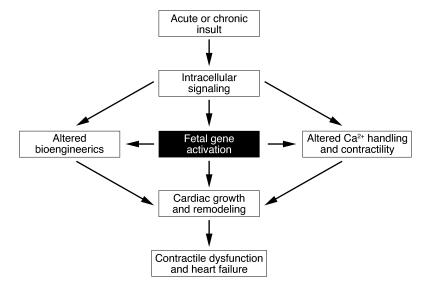
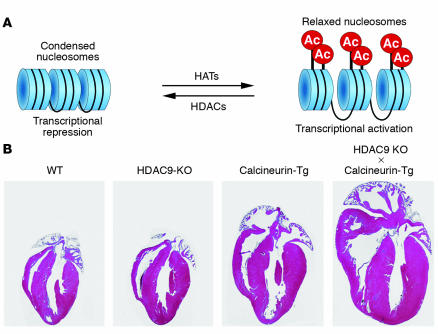
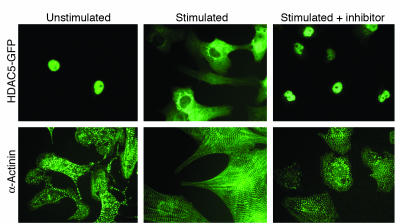
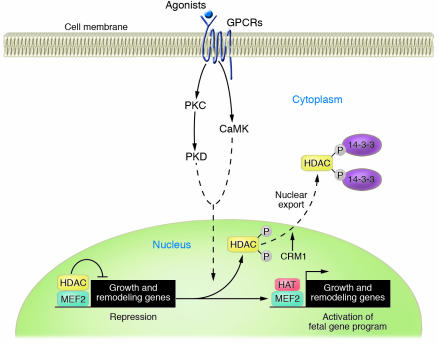
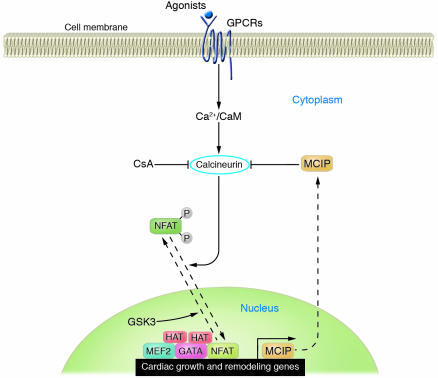
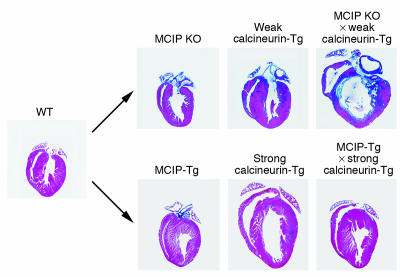
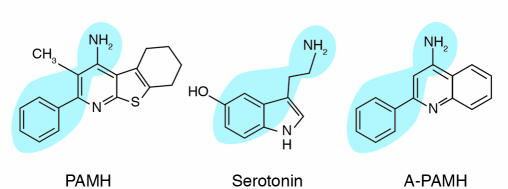
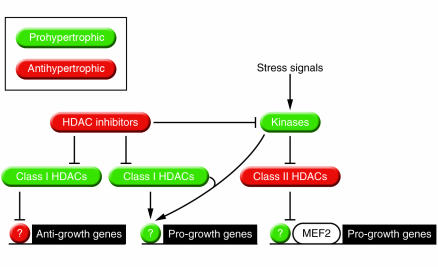
In response to acute and chronic stresses, the heart frequently undergoes a remodeling process that is accompanied by myocyte hypertrophy, impaired contractility, and pump failure, often culminating in sudden death. The existence of redundant signaling pathways that trigger heart failure poses challenges for therapeutic intervention. Cardiac remodeling is associated with the activation of a pathological gene program that weakens cardiac performance. Thus, targeting the disease process at the level of gene expression represents a potentially powerful therapeutic approach. In this review, we describe strategies for normalizing gene expression in the failing heart with small molecules that control signal transduction pathways directed at transcription factors and associated chromatin-modifying enzymes.
Figures
References
-
- Linseman JV, Bristow MR. Drug therapy and heart failure prevention. Circulation. 2003;107:1234–1236. - PubMed
-
- Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am. Heart J. 2001;141:334–341. - PubMed
-
- Okin PM, et al. for the LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA. 2004;292:2343–2349. - PubMed
-
- Devereux RB, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–2356. - PubMed
-
- Gardin JM, Lauer MS. Left ventricular hypertrophy: the next treatable, silent killer? JAMA. 2004;292:2396–2398. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
