

Review
doi: 10.1172/JCI24159.
Altered intracellular Ca2+ handling in heart failure
Affiliations
- PMID: 15765137
- PMCID: PMC1052007
- DOI: 10.1172/JCI24159
Item in Clipboard
Review
Altered intracellular Ca2+ handling in heart failure
J Clin Invest.
2005 Mar.
Abstract
Structural and functional alterations in the Ca2+ regulatory proteins present in the sarcoplasmic reticulum have recently been shown to be strongly involved in the pathogenesis of heart failure. Chronic activation of the sympathetic nervous system or of the renin-angiotensin system induces abnormalities in both the function and structure of these proteins. We review here the considerable body of evidence that has accumulated to support the notion that such abnormalities contribute to a defectiveness of contractile performance and hence to the progression of heart failure.
Figures
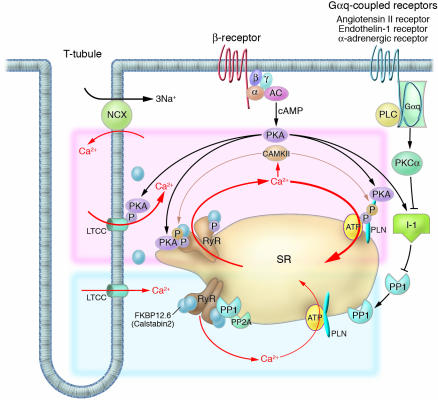
Intracellular Ca2+ cycling and associated signaling pathway in cardiomyocytes. On a beat-by-beat basis, a calcium transient is evoked by the initial influx of a small amount of Ca2+ through the LTCC and the subsequent large-scale Ca2+ release from the SR through the RyR. During diastole, cytosolic Ca2+ is taken up into the SR by the PLN-regulated SERCA2a pump. β receptor–mediated PKA stimulation regulates this Ca2+ cycling by phosphorylating LTCC, RyR, and PLN. In normal hearts, sympathetic stimulation activates β1-adrenergic receptor, which in turn stimulates the production of cAMP by adenylyl cyclase and thereby activates PKA. PKA phosphorylates PLN and RyR, both of which contribute to an increased intracellular Ca2+ transient and enhanced cellular contractility (pink zone signal). PP1 and PP2A regulate the dephosphorylation process of these Ca2+ regulatory proteins (RyR, PLN, LTCC) (blue zone signaling). Activation of the Gαq-coupled receptors (angiotensin II receptor, endothelin 1 receptor, or α-adrenergic receptor) activates PLC, which in turn activates PKC-α. The PKC-α phosphorylates I-1, augmenting the activity of PP1 and causing hypophosphorylation of PLB. The PLB hypophosphorylation inhibits SERCA2a activity, thereby decreasing SR Ca2+ uptake. The increased Ca2+ level in the cytosol activates CAMKII, which affects the functions of RyR and PLN. Activation or deactivation of these molecules at a node in the signaling cascade affects beat-by-beat Ca2+ cycling, and such maneuvers have recently been highlighted as potential new therapeutic strategies against HF. α, G protein subunit α; β, G protein subunit β; γ, G protein subunit γ; AC, adenylyl cyclase; PLC, phospholipase C.
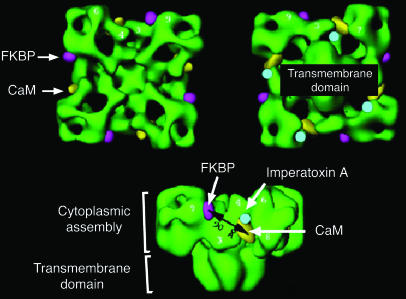
Three-dimensional structure of the skeletal muscle RyR, with some key sites of protein interactions. FKBP, FK506-binding protein; CaM, calmodulin. Image reprinted with permission from the Journal of Biological Chemistry (115).
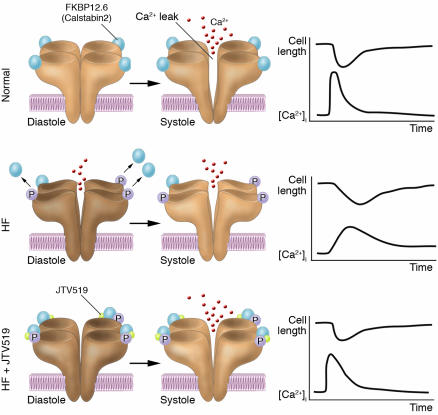
Therapeutic strategy involving FKBP12.6-mediated stabilization of RyR. A small influx of Ca2+ through the LTCC leads to the release of a large amount of Ca2+ from the SR through RyR in the normal heart. In HF, however, PKA-mediated hyperphosphorylation of RyR2 occurs, and this in turn dissociates FKBP12.6 from RyR2, leading to a diastolic Ca2+ leak through RyR2. This results in the Ca2+ transient being diminished (due to the reduced SR Ca2+ content and dyssynchronous Ca2+ release). Administration of a new compound, the 1,4-benzothiazepine derivative JTV519, normalizes this abnormal channel gating by restoring the conformational state of RyR and by rebinding FKBP12.6 to the channel complex. Thereby, JTV519 normalizes Ca2+ cycling and contractile function in failing cardiac myocytes and hence provides chronic suppression of progressive left ventricular dysfunction in HF. P, PKA phosphorylation at serine 2809; [Ca2+]i, intracellular [Ca2+].
References
-
- Francis GS. Pathophysiology of chronic heart failure. Am. J. Med. 2001;110:37S–46S. - PubMed
-
- Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation. 2000;102:IV14–IV23. - PubMed
-
- Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J. Mol. Cell. Cardiol. 2002;34:951–969. - PubMed
-
- Meyer M, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–784. - PubMed
-
- Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ. Res. 2003;92:350–358. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
