

Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus
- PMID: 15767435
- PMCID: PMC1061535
- DOI: 10.1128/JVI.79.7.4347-4356.2005
Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus
Abstract
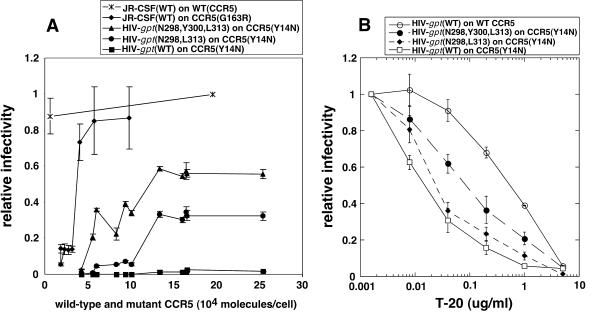
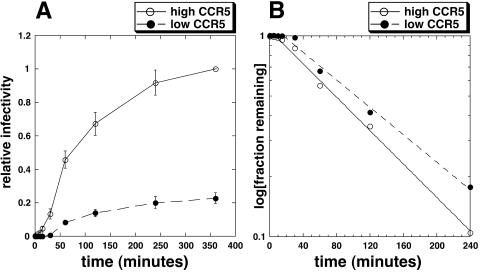
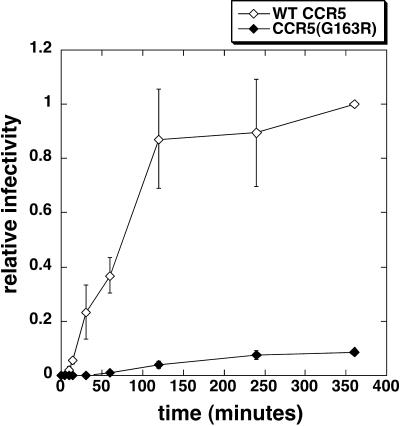
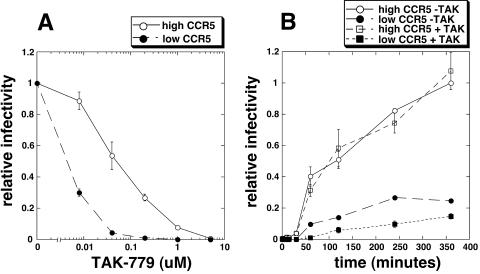
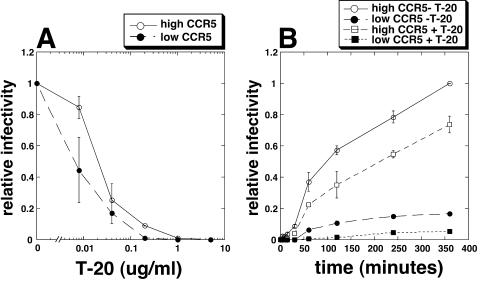
Replication of human immunodeficiency virus type 1 (HIV-1) in diverse conditions limiting for viral entry into cells frequently leads to adaptive mutations in the V3 loop of the gp120 envelope glycoprotein. This has suggested that the V3 loop limits the efficiencies of HIV-1 infections, possibly by directly affecting gp120-coreceptor affinities. In contrast, V3 loop mutations that enable HIV-1(JR-CSF) to use the low-affinity mutant coreceptor CCR5(Y14N) are shown here to have negligible effects on the virus-coreceptor affinity but to dramatically accelerate the irreversible conformational conversion of the envelope gp41 subunits from a three-stranded coil into a six-helix bundle. This slow step is blocked irreversibly by the inhibitor T-20. To further evaluate the role of entry rates in controlling infection efficiencies and viral adaptations, we developed methods to quantitatively measure viral entry kinetics. The virions were adsorbed by spinoculation at 4 degrees C onto HeLa-CD4/CCR5 cell clones that either had limiting or saturating concentrations of CCR5. After warming to 37 degrees C, the completion of entry was monitored over time by the resistance of infections to the competitive CCR5 inhibitor TAK-779. Our results suggest that the efficiency of entry of cell-attached infectious HIV-1 is principally controlled by three kinetic processes. The first is a lag phase that is caused in part by the concentration-dependent reversible association of virus with CD4 and CCR5 to form an equilibrium assemblage of complexes. Second, this assembly step lowers but does not eliminate a large activation energy barrier for a rate-limiting, CCR5-dependent conformational change in gp41 that is sensitive to blockage by T-20. The rate of infection therefore depends on the fraction of infectious virions that are sufficiently saturated with CCR5 to undergo this conformational change and on the magnitude of the activation energy barrier. Although only a small fraction of fully assembled viral complexes overcome this barrier per hour, the ensuing steps of entry are rapidly completed within 5 to 10 min. Thus, this barrier limits the overall flow rate at which the attached virions enter cells, but it has no effect on the lag time that precedes this entry flow. Third, a relatively rapid and kinetically dominant process of viral inactivation, which may partly involve endocytosis, competes with infectious viral entry. Our results suggest that the V3 loop of gp120 has a major effect on the rate-limiting coreceptor-dependent conformational change in gp41 and that adaptive viral mutations, including V3 loop mutations, function kinetically by accelerating this inherently slow step in the entry pathway.
Figures
References
-
- Baba, M., O. Nishimura, N. Kanzaki, M. Okamoto, H. Sawada, Y. Iizawa, M. Shiraishi, Y. Aramaki, K. Okonogi, Y. Ogawa, K. Meguro, and M. Fujino. 1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. USA 96:5698-5703. - PMC - PubMed
-
- Berger, E. A., P. M. Murphy, and J. M. Farber. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17:657-700. - PubMed
-
- Cavrois, M., C. De Noronha, and W. C. Greene. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20:1151-1154. - PubMed
-
- Cavrois, M., J. Neidleman, M. Bigos, and W. C. Greene. 2004. Fluorescence resonance energy transfer-based HIV-1 virion fusion assay. Methods Mol. Biol. 263:333-344. - PubMed
-
- Chan, D. C., and P. S. Kim. 1998. HIV entry and its inhibition. Cell 93:681-684. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
