

Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins
- PMID: 15767576
- PMCID: PMC555489
- DOI: 10.1073/pnas.0407499102
Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins
Abstract
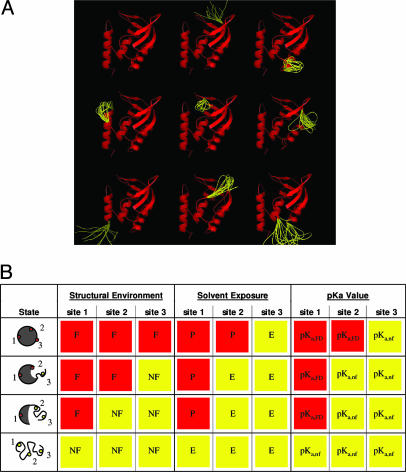
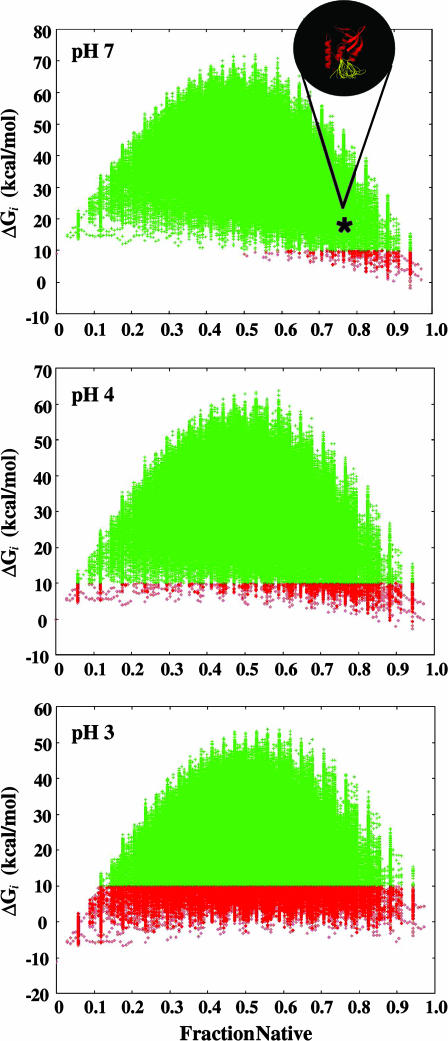
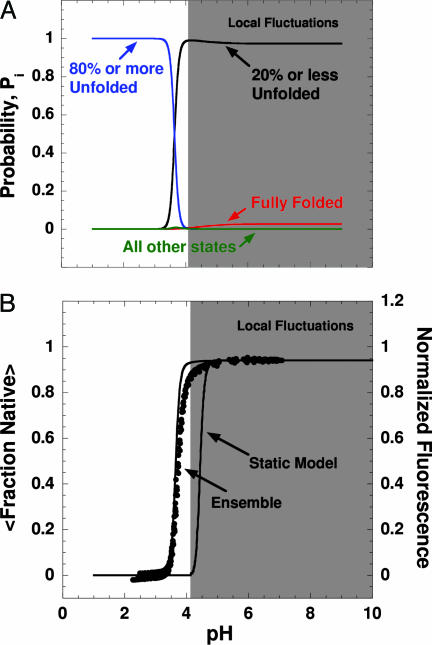
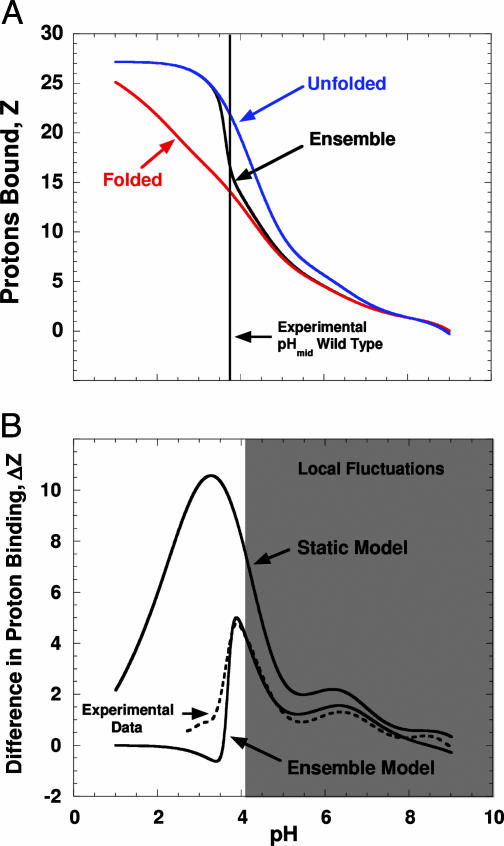
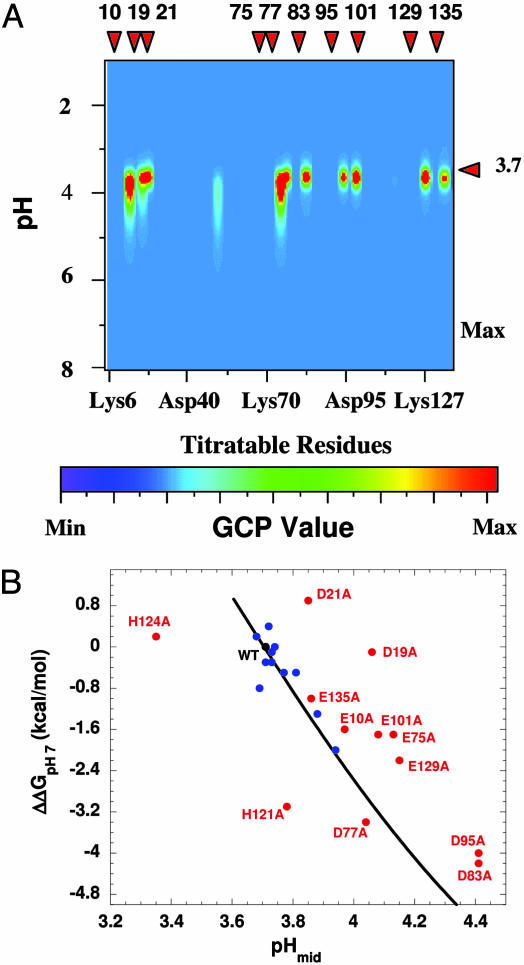
Local conformational fluctuations in proteins can affect the coupling between ligand binding and global structural transitions. This finding was established by monitoring quantitatively how the population distribution in the ensemble of microstates of staphylococcal nuclease was affected by proton binding. Analysis of acid unfolding and proton-binding data with an ensemble-based model suggests that local fluctuations: (i) can be effective modulators of ligand-binding affinities, (ii) are important determinants of the cooperativity of ligand-driven global structural transitions, and (iii) are well represented thermodynamically as local unfolding processes. These studies illustrate how an ensemble-based description of proteins can be used to describe quantitatively the interdependence of local conformational fluctuations, ligand-binding processes, and global structural transitions. This level of understanding of the relationship between conformation, energy, and dynamics is required for a detailed mechanistic understanding of allostery, cooperativity, and other complex functional and regulatory properties of macromolecules.
Figures
Similar articles
-
Ligand effects on the protein ensemble: unifying the descriptions of ligand binding, local conformational fluctuations, and protein stability.Methods Cell Biol. 2008;84:871-91. doi: 10.1016/S0091-679X(07)84027-1. Methods Cell Biol. 2008. PMID: 17964952
-
Molecular mechanisms of pH-driven conformational transitions of proteins: insights from continuum electrostatics calculations of acid unfolding.Proteins. 2006 Apr 1;63(1):113-26. doi: 10.1002/prot.20797. Proteins. 2006. PMID: 16400648
-
pH dependence of conformational fluctuations of the protein backbone.Proteins. 2014 Nov;82(11):3132-43. doi: 10.1002/prot.24673. Epub 2014 Sep 4. Proteins. 2014. PMID: 25137073
-
Staphylococcal nuclease: a showcase of m-value effects.Adv Protein Chem. 1995;46:217-47. doi: 10.1016/s0065-3233(08)60336-8. Adv Protein Chem. 1995. PMID: 7771319 Review. No abstract available.
-
Staphylococcal nuclease reviewed: a prototypic study in contemporary enzymology. IV. The nuclease as a model for protein folding.Mol Cell Biochem. 1979 Feb 9;23(3):131-41. doi: 10.1007/BF00219452. Mol Cell Biochem. 1979. PMID: 90333 Review.
Cited by
-
Rapid calculation of protein pKa values using Rosetta.Biophys J. 2012 Aug 8;103(3):587-595. doi: 10.1016/j.bpj.2012.06.044. Biophys J. 2012. PMID: 22947875 Free PMC article.
-
Identifying allosteric fluctuation transitions between different protein conformational states as applied to Cyclin Dependent Kinase 2.BMC Bioinformatics. 2007 Feb 7;8:45. doi: 10.1186/1471-2105-8-45. BMC Bioinformatics. 2007. PMID: 17286863 Free PMC article.
-
Interaction of the new inhibitor paxlovid (PF-07321332) and ivermectin with the monomer of the main protease SARS-CoV-2: A volumetric study based on molecular dynamics, elastic networks, classical thermodynamics and SPT.Comput Biol Chem. 2022 Aug;99:107692. doi: 10.1016/j.compbiolchem.2022.107692. Epub 2022 May 14. Comput Biol Chem. 2022. PMID: 35640480 Free PMC article.
-
Local packing modulates diversity of iron pathways and cooperative behavior in eukaryotic and prokaryotic ferritins.J Chem Phys. 2014 Mar 21;140(11):115104. doi: 10.1063/1.4868229. J Chem Phys. 2014. PMID: 24655206 Free PMC article.
-
Denatured-state energy landscapes of a protein structural database reveal the energetic determinants of a framework model for folding.J Mol Biol. 2008 Sep 19;381(5):1184-201. doi: 10.1016/j.jmb.2008.06.046. Epub 2008 Jun 24. J Mol Biol. 2008. PMID: 18616947 Free PMC article.
References
-
- Englander, S. W. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 213–238. - PubMed
-
- Swint-Kruse, L. & Robertson, A. D. (1996) Biochemistry 35, 171–180. - PubMed
-
- Hvidt, A. & Nielsen, S. O. (1966) Adv. Protein Chem. 21, 287–386. - PubMed
-
- Chamberlain, A. K., Handel. T. & Marqusee, S. (1996) Nat. Struct. Biol. 3, 782–787. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
