

An initial strategy for the systematic identification of functional elements in the human genome by low-redundancy comparative sequencing
- PMID: 15778292
- PMCID: PMC555705
- DOI: 10.1073/pnas.0409882102
An initial strategy for the systematic identification of functional elements in the human genome by low-redundancy comparative sequencing
Abstract
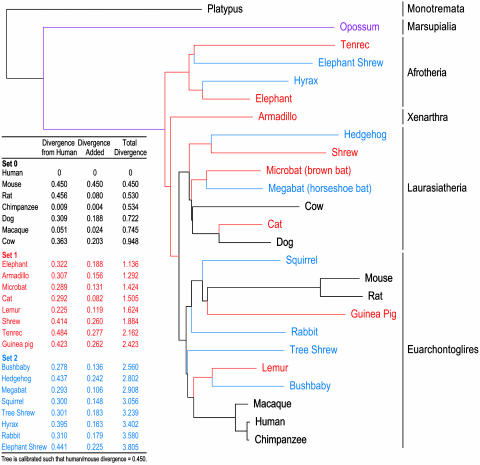
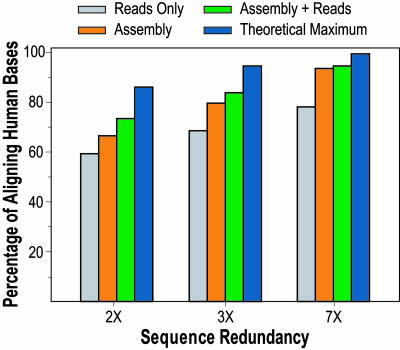
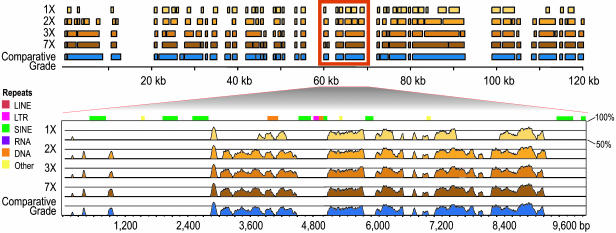
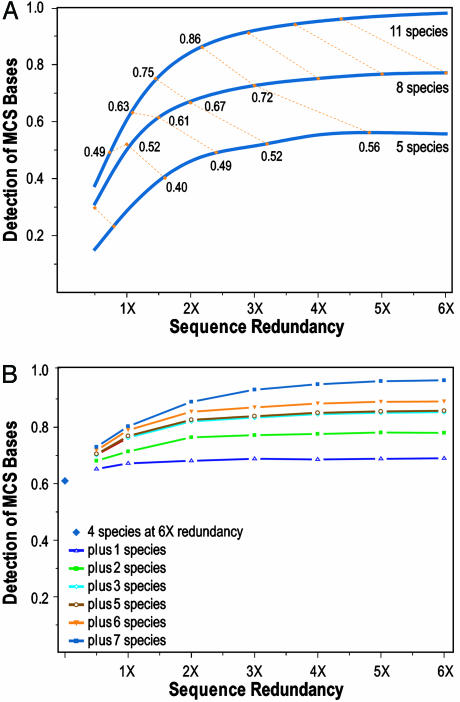
With the recent completion of a high-quality sequence of the human genome, the challenge is now to understand the functional elements that it encodes. Comparative genomic analysis offers a powerful approach for finding such elements by identifying sequences that have been highly conserved during evolution. Here, we propose an initial strategy for detecting such regions by generating low-redundancy sequence from a collection of 16 eutherian mammals, beyond the 7 for which genome sequence data are already available. We show that such sequence can be accurately aligned to the human genome and used to identify most of the highly conserved regions. Although not a long-term substitute for generating high-quality genomic sequences from many mammalian species, this strategy represents a practical initial approach for rapidly annotating the most evolutionarily conserved sequences in the human genome, providing a key resource for the systematic study of human genome function.
Figures
References
-
- Collins, F. S., Green, E. D., Guttmacher, A. E. & Guyer, M. S. (2003) Nature 422, 835-847. - PubMed
-
- Ohta, T. (1976) Nature 263, 74-76. - PubMed
-
- International Mouse Genome Sequencing Consortium (2002) Nature 420, 520-562. - PubMed
-
- Rat Genome Sequencing Project Consortium (2004) Nature 428, 493-521. - PubMed
-
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S. & Haussler, D. (2004) Science 304, 1321-1325. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
