

The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages
- PMID: 15788529
- PMCID: PMC556006
- DOI: 10.1073/pnas.0501140102
The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages
Abstract
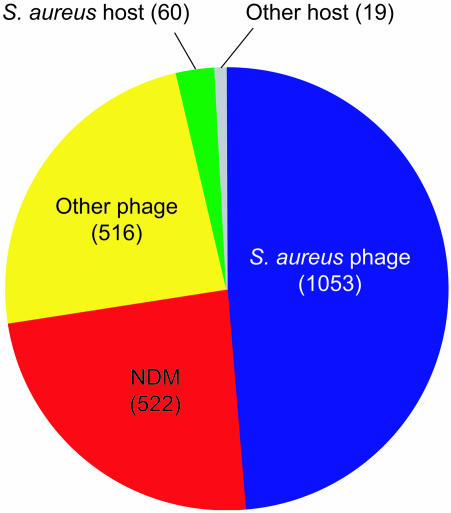
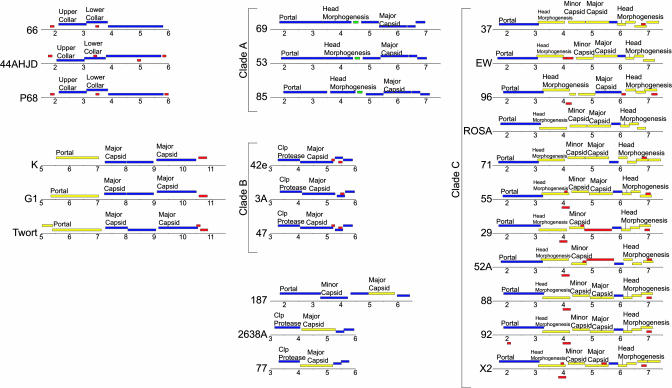
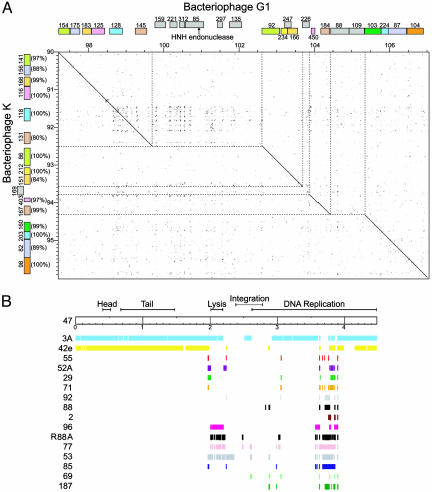
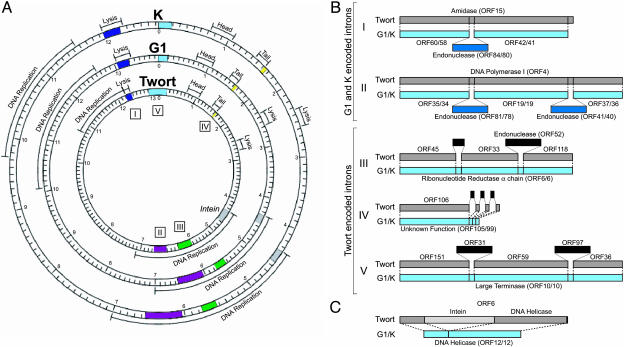
Bacteriophages are the most abundant life forms in the biosphere. They play important roles in bacterial ecology, evolution, adaptation to new environments, and pathogenesis of human bacterial infections. Here, we report the complete genomic sequences, and predicted proteins of 27 bacteriophages of the Gram-positive bacterium Staphylococcus aureus. Comparative nucleotide and protein sequence analysis indicates that these phages are a remarkable source of untapped genetic diversity, encoding 2,170 predicted protein-encoding ORFs, of which 1,402 cannot be annotated for structure or function, and 522 are proteins with no similarity to other phage or bacterial sequences. Based on their genome size, organization of their gene map and comparative nucleotide and protein sequence analysis, the S. aureus phages can be organized into three groups. Comparison of their gene maps reveals extensive genome mosaicism, hinting to a large reservoir of unidentified S. aureus phage genes. Among the phages in the largest size class (178-214 kbp) that we characterized is phage Twort, the first discovered bacteriophage (responsible for the Twort-D'Herelle effect). These phage genomes offer an exciting opportunity to discern molecular mechanisms of phage evolution and diversity.
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
