

Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage
- PMID: 15788536
- PMCID: PMC555986
- DOI: 10.1073/pnas.0408595102
Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage
Abstract
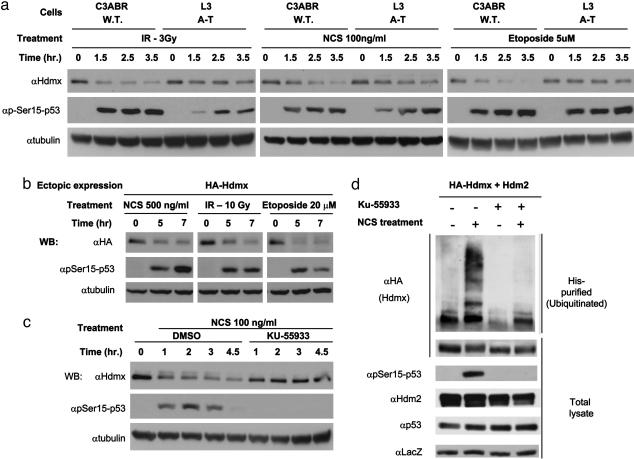
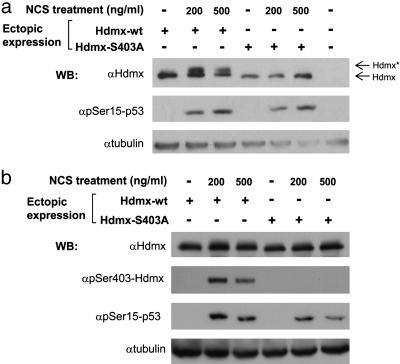
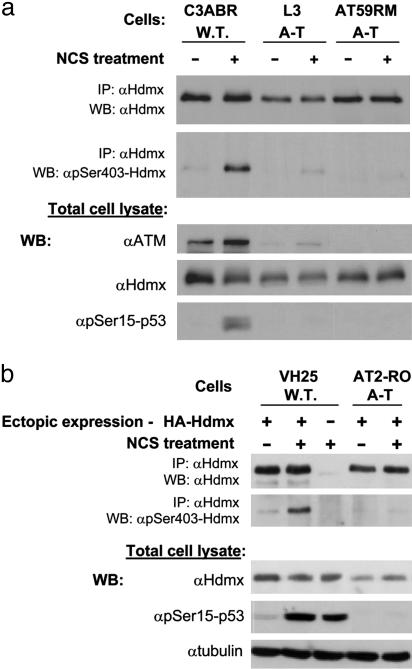
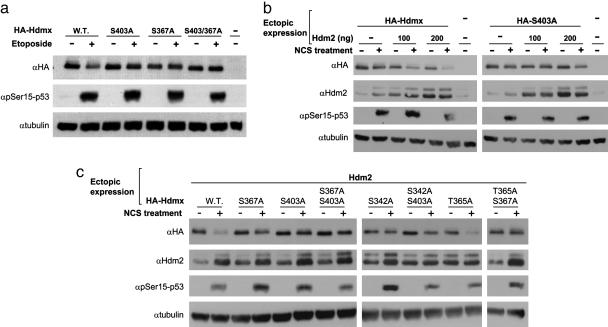
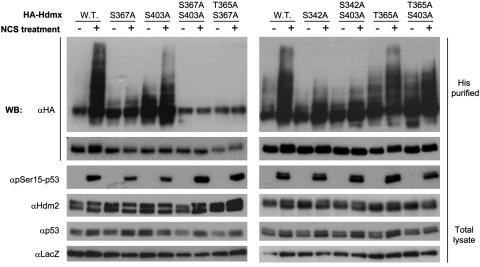
Maintenance of genomic stability depends on the DNA damage response, an extensive signaling network that is activated by DNA lesions such as double-strand breaks (DSBs). The primary activator of the mammalian DSB response is the nuclear protein kinase ataxia-telangiectasia, mutated (ATM), which phosphorylates key players in various arms of this network. The activation and stabilization of the p53 protein play a major role in the DNA damage response and are mediated by ATM-dependent posttranslational modifications of p53 and Mdm2, a ubiquitin ligase of p53. p53's response to DNA damage also depends on Mdm2-dependent proteolysis of Mdmx, a homologue of Mdm2 that represses p53's transactivation function. Here we show that efficient damage-induced degradation of human Hdmx depends on functional ATM and at least three sites on the Hdmx that are phosphorylated in response to DSBs. One of these sites, S403, is a direct ATM target. Accordingly, each of these sites is important for Hdm2-mediated ubiquitination of Hdmx after DSB induction. These results demonstrate a sophisticated mechanism whereby ATM fine-tunes the optimal activation of p53 by simultaneously modifying each player in the process.
Figures
Similar articles
-
Differential roles of ATM- and Chk2-mediated phosphorylations of Hdmx in response to DNA damage.Mol Cell Biol. 2006 Sep;26(18):6819-31. doi: 10.1128/MCB.00562-06. Mol Cell Biol. 2006. PMID: 16943424 Free PMC article.
-
ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage.EMBO J. 2005 Oct 5;24(19):3411-22. doi: 10.1038/sj.emboj.7600812. Epub 2005 Sep 15. EMBO J. 2005. PMID: 16163388 Free PMC article.
-
ATM engages autodegradation of the E3 ubiquitin ligase COP1 after DNA damage.Science. 2006 Aug 25;313(5790):1122-6. doi: 10.1126/science.1127335. Science. 2006. PMID: 16931761
-
The ATM-dependent DNA damage signaling pathway.Cold Spring Harb Symp Quant Biol. 2005;70:99-109. doi: 10.1101/sqb.2005.70.002. Cold Spring Harb Symp Quant Biol. 2005. PMID: 16869743 Review.
-
How to activate p53.Curr Biol. 2000 Apr 20;10(8):R315-7. doi: 10.1016/s0960-9822(00)00439-5. Curr Biol. 2000. PMID: 10801407 Review.
Cited by
-
MDM2 oligomers: antagonizers of the guardian of the genome.Oncogene. 2016 Dec 1;35(48):6157-6165. doi: 10.1038/onc.2016.88. Epub 2016 Apr 4. Oncogene. 2016. PMID: 27041565 Free PMC article. Review.
-
USP7 Is a Master Regulator of Genome Stability.Front Cell Dev Biol. 2020 Aug 5;8:717. doi: 10.3389/fcell.2020.00717. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32850836 Free PMC article. Review.
-
Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant.PLoS One. 2012;7(6):e39956. doi: 10.1371/journal.pone.0039956. Epub 2012 Jun 29. PLoS One. 2012. PMID: 22768182 Free PMC article.
-
β-TrCP1 facilitates cell cycle checkpoint activation, DNA repair, and cell survival through ablation of β-TrCP2 in response to genotoxic stress.J Biol Chem. 2021 Jan-Jun;296:100511. doi: 10.1016/j.jbc.2021.100511. Epub 2021 Mar 4. J Biol Chem. 2021. PMID: 33676897 Free PMC article.
-
The MDMX Acidic Domain Uses Allovalency to Bind Both p53 and MDMX.J Mol Biol. 2022 Nov 30;434(22):167844. doi: 10.1016/j.jmb.2022.167844. Epub 2022 Sep 29. J Mol Biol. 2022. PMID: 36181774 Free PMC article.
References
-
- Norbury, C. J. & Hickson, I. D. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 367–401. - PubMed
-
- Shiloh, Y. (2003) Nat. Rev. Cancer 3, 155–168. - PubMed
-
- Bakkenist, C. J. & Kastan, M. B. (2004) Cell 118, 9–17. - PubMed
-
- Jackson, S. P. (2002) Carcinogenesis 23, 687–696. - PubMed
-
- Bassing, C. H. & Alt, F. W. (2004) DNA Repair (Amsterdam) 3, 781–796. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
