

Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis
- PMID: 15793702
- PMCID: PMC1199364
- DOI: 10.1086/429417
Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis
Abstract
P450 oxidoreductase (POR) is the obligatory flavoprotein intermediate that transfers electrons from reduced nicotinamide adenine dinucleotide phosphate (NADPH) to all microsomal cytochrome P450 enzymes. Although mouse Por gene ablation causes embryonic lethality, POR missense mutations cause disordered steroidogenesis, ambiguous genitalia, and Antley-Bixler syndrome (ABS), which has also been attributed to fibroblast growth factor receptor 2 (FGFR2) mutations. We sequenced the POR gene and FGFR2 exons 8 and 10 in 32 individuals with ABS and/or hormonal findings that suggested POR deficiency. POR and FGFR2 mutations segregated completely. Fifteen patients carried POR mutations on both alleles, 4 carried mutations on only one allele, 10 carried FGFR2 or FGFR3 mutations, and 3 patients carried no mutations. The 34 affected POR alleles included 10 with A287P (all from whites) and 7 with R457H (four Japanese, one African, two whites); 17 of the 34 alleles carried 16 "private" mutations, including 9 missense and 7 frameshift mutations. These 11 missense mutations, plus 10 others found in databases or reported elsewhere, were recreated by site-directed mutagenesis and were assessed by four assays: reduction of cytochrome c, oxidation of NADPH, support of 17alpha-hydroxylase activity, and support of 17,20 lyase using human P450c17. Assays that were based on cytochrome c, which is not a physiologic substrate for POR, correlated poorly with clinical phenotype, but assays that were based on POR's support of catalysis by P450c17--the enzyme most closely associated with the hormonal phenotype--provided an excellent genotype/phenotype correlation. Our large survey of patients with ABS shows that individuals with an ABS-like phenotype and normal steroidogenesis have FGFR mutations, whereas those with ambiguous genitalia and disordered steroidogenesis should be recognized as having a distinct new disease: POR deficiency.
Figures
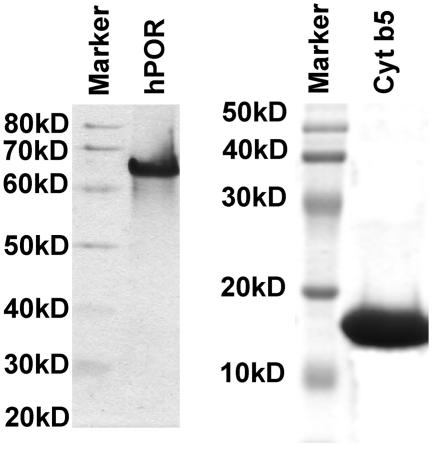
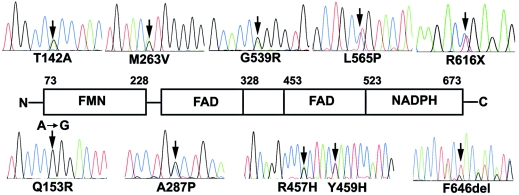
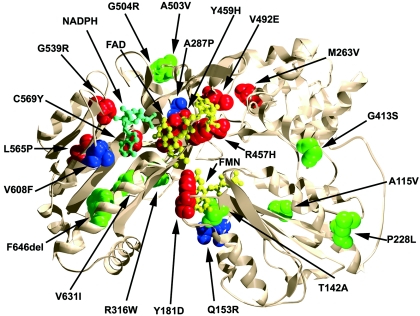

Comment in
-
Use of the term "Antley-Bixler syndrome": minimizing confusion.Am J Hum Genet. 2005 Aug;77(2):327-8; author reply 328-40. doi: 10.1086/432164. Am J Hum Genet. 2005. PMID: 16145814 Free PMC article. No abstract available.
References
Electronic-Database Information
-
- Berkeley Drosophila Genome Project, http://www.fruitfly.org/seq_tools/splice.html (for the program NNSplice)
-
- BioVentures, http://www.bioventures.com/products/dmg/por/ (for POR gene polymorphism data)
-
- Centre National de la Recherche Scientifique Marseilles Group in Enzyme Kinetics and Control Analysis, http://bip.cnrs-mrs.fr/bip10/homepage.htm (for the program LEONORA)
-
- ClustalW, http://www.ebi.ac.uk/clustalw/index.html
-
- DeepView, http://www.expasy.org/spdbv/
References
-
- Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A (2004) Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am J Med Genet A 128A:333–339 - PubMed
-
- Aleck KA, Bartley DL (1997) Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet 72:253–256 - PubMed
-
- Antley R, Bixler D (1975) Trapezoidocephaly, midfacial hypoplasia and cartilage abnormalities with multiple synostoses and skeletal fractures. Birth Defects Orig Artic Ser 11:397–401 - PubMed
-
- Arlt W, Auchus RJ, Miller WL (2001) Thiazolidinediones but not metformin directly inhibit the steroidogenic enzymes P450c17 and 3β-hydroxysteroid dehydrogenase. J Biol Chem 276:16767–16771 - PubMed
-
- Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EM, Stewart PM, Shackleton CH (2004) Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 363:2128–2135 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
