

Modeling gene and genome duplications in eukaryotes
- PMID: 15800040
- PMCID: PMC556253
- DOI: 10.1073/pnas.0501102102
Modeling gene and genome duplications in eukaryotes
Abstract
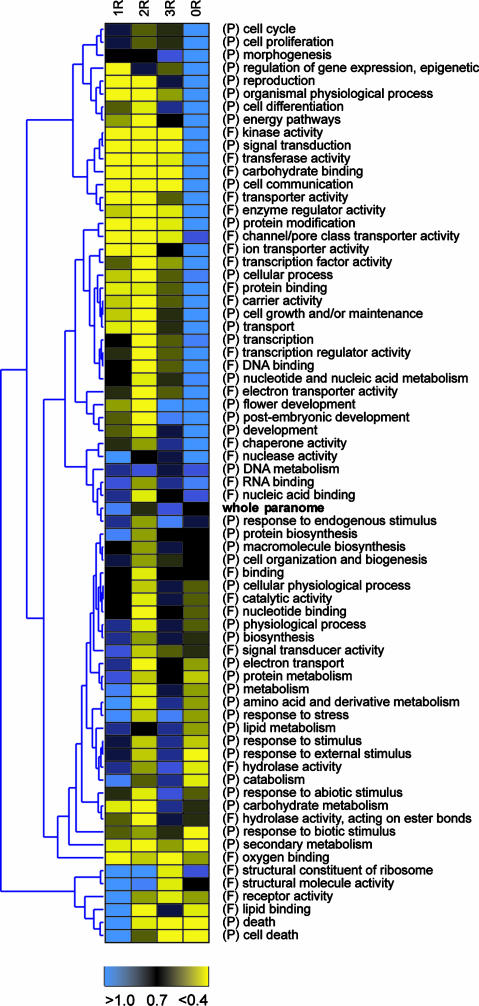
Recent analysis of complete eukaryotic genome sequences has revealed that gene duplication has been rampant. Moreover, next to a continuous mode of gene duplication, in many eukaryotic organisms the complete genome has been duplicated in their evolutionary past. Such large-scale gene duplication events have been associated with important evolutionary transitions or major leaps in development and adaptive radiations of species. Here, we present an evolutionary model that simulates the duplication dynamics of genes, considering genome-wide duplication events and a continuous mode of gene duplication. Modeling the evolution of the different functional categories of genes assesses the importance of different duplication events for gene families involved in specific functions or processes. By applying our model to the Arabidopsis genome, for which there is compelling evidence for three whole-genome duplications, we show that gene loss is strikingly different for large-scale and small-scale duplication events and highly biased toward certain functional classes. We provide evidence that some categories of genes were almost exclusively expanded through large-scale gene duplication events. In particular, we show that the three whole-genome duplications in Arabidopsis have been directly responsible for >90% of the increase in transcription factors, signal transducers, and developmental genes in the last 350 million years. Our evolutionary model is widely applicable and can be used to evaluate different assumptions regarding small- or large-scale gene duplication events in eukaryotic genomes.
Figures



References
-
- Ohno, S. (1970) Evolution by Gene Duplication (Springer, New York).
-
- Lynch, M. & Conery, J. S. (2000) Science 290, 1151–1155. - PubMed
-
- Lynch, M. & Conery, J. S. (2003) J. Struct. Funct. Genomics 3, 35–44. - PubMed
-
- Li, W.-H., Gu, Z., Cavalcanti, A. R. O. & Nekrutenko, A. (2003) J. Struct. Funct. Genomics 3, 27–34. - PubMed
-
- Wolfe, K. H. (2001) Nat. Rev. Genet. 2, 333–341. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
