

ECgene: genome-based EST clustering and gene modeling for alternative splicing
- PMID: 15805497
- PMCID: PMC1074371
- DOI: 10.1101/gr.3030405
ECgene: genome-based EST clustering and gene modeling for alternative splicing
Abstract
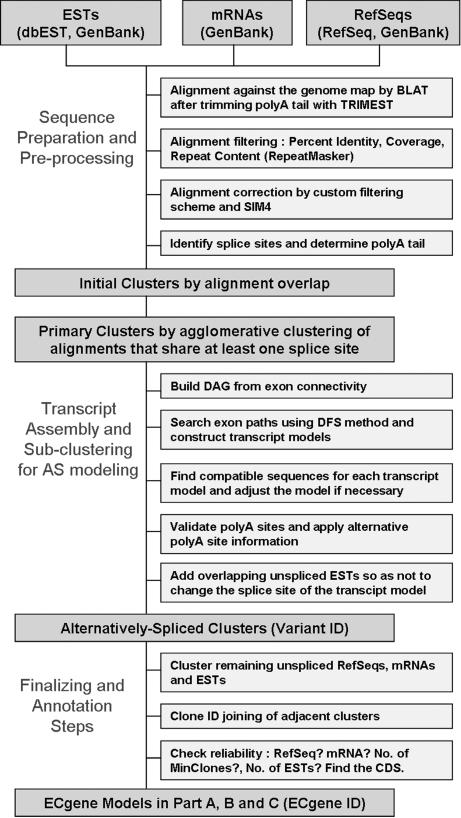
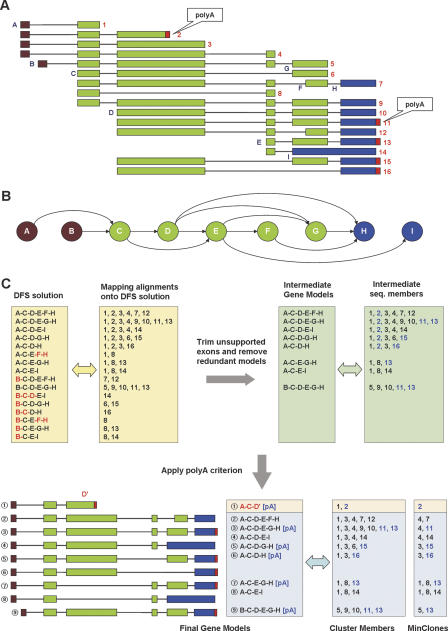
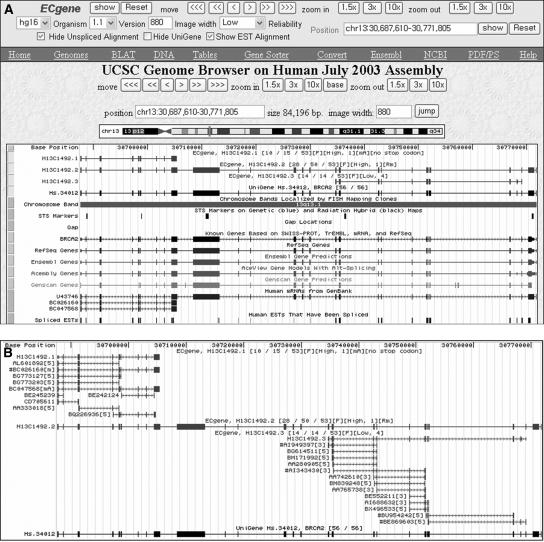
With the availability of the human genome map and fast algorithms for sequence alignment, genome-based EST clustering became a viable method for gene modeling. We developed a novel gene-modeling method, ECgene (Gene modeling by EST Clustering), which combines genome-based EST clustering and the transcript assembly procedure in a coherent and consistent fashion. Specifically, ECgene takes alternative splicing events into consideration. The position of splice sites (i.e., exon-intron boundaries) in the genome map is utilized as the critical information in the whole procedure. Sequences that share any splice sites are grouped together to define an EST cluster in a manner similar to that of the genome-based version of the UniGene algorithm. Transcript assembly is achieved using graph theory that represents the exon connectivity in each cluster as a directed acyclic graph (DAG). Distinct paths along exons correspond to possible gene models encompassing all alternative splicing events. EST sequences in each cluster are subclustered further according to the compatibility with gene structure of each splice variant, and they can be regarded as clone evidence for the corresponding isoform. The reliability of each isoform is assessed from the nature of cluster members and from the minimum number of clones required to reconstruct all exons in the transcript.
Figures
Similar articles
-
ASmodeler: gene modeling of alternative splicing from genomic alignment of mRNA, EST and protein sequences.Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W181-6. doi: 10.1093/nar/gkh404. Nucleic Acids Res. 2004. PMID: 15215376 Free PMC article.
-
ECgene: genome annotation for alternative splicing.Nucleic Acids Res. 2005 Jan 1;33(Database issue):D75-9. doi: 10.1093/nar/gki118. Nucleic Acids Res. 2005. PMID: 15608289 Free PMC article.
-
ECgene: an alternative splicing database update.Nucleic Acids Res. 2007 Jan;35(Database issue):D99-103. doi: 10.1093/nar/gkl992. Epub 2006 Nov 28. Nucleic Acids Res. 2007. PMID: 17132829 Free PMC article.
-
How prevalent is functional alternative splicing in the human genome?Trends Genet. 2004 Feb;20(2):68-71. doi: 10.1016/j.tig.2003.12.004. Trends Genet. 2004. PMID: 14746986 Review.
-
Computational methods for alternative splicing prediction.Brief Funct Genomic Proteomic. 2006 Mar;5(1):46-51. doi: 10.1093/bfgp/ell011. Epub 2006 Feb 20. Brief Funct Genomic Proteomic. 2006. PMID: 16769678 Review.
Cited by
-
The ASAP II database: analysis and comparative genomics of alternative splicing in 15 animal species.Nucleic Acids Res. 2007 Jan;35(Database issue):D93-8. doi: 10.1093/nar/gkl884. Epub 2006 Nov 15. Nucleic Acids Res. 2007. PMID: 17108355 Free PMC article.
-
Evolution of alternative splicing after gene duplication.Genome Res. 2006 Feb;16(2):182-9. doi: 10.1101/gr.4197006. Epub 2005 Dec 19. Genome Res. 2006. PMID: 16365379 Free PMC article.
-
Modeling transcriptome based on transcript-sampling data.PLoS One. 2008 Feb 20;3(2):e1659. doi: 10.1371/journal.pone.0001659. PLoS One. 2008. PMID: 18286206 Free PMC article.
-
ASPIC: a novel method to predict the exon-intron structure of a gene that is optimally compatible to a set of transcript sequences.BMC Bioinformatics. 2005 Oct 5;6:244. doi: 10.1186/1471-2105-6-244. BMC Bioinformatics. 2005. PMID: 16207377 Free PMC article.
-
Characterising alternate splicing and tissue specific expression in the chicken from ESTs.Cytogenet Genome Res. 2007;117(1-4):268-77. doi: 10.1159/000103188. Cytogenet Genome Res. 2007. PMID: 17675868 Free PMC article.
References
-
- Adams, M.D., Kelley, J.M., Gocayne, J.D., Dubnick, M., Polymeropoulos, M.H., Xiao, H., Merril, C.R., Wu, A., Olde, B., Moreno, R.F., et al. 1991. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 252: 1651-1656. - PubMed
-
- Black, D.L. 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. BioChem. 72: 291-336. - PubMed
-
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. - PubMed
-
- Caceres, J.F. and Kornblihtt, A.R. 2002. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 18: 186-193. - PubMed
Web site references
-
- http://genome.ewha.ac.kr/ECgene; ECgene Web site.
-
- ftp://ftp.ncbi.nlm.nih.gov/genbank/; GenBank FTP site.
-
- ftp://hgdownload.cse.ucsc.edu/goldenPath/; Genome Browser FTP site at the UCSC Genome Center.
-
- ftp:/ftp.ncbi.nlm.nih.gov/refseq/release/; RefSeq FTP site.
-
- http://genome.ucsc.edu; UCSC Genome Bioinformatics Home.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials