

Closing in on the C. elegans ORFeome by cloning TWINSCAN predictions
- PMID: 15805498
- PMCID: PMC1074372
- DOI: 10.1101/gr.3329005
Closing in on the C. elegans ORFeome by cloning TWINSCAN predictions
Abstract
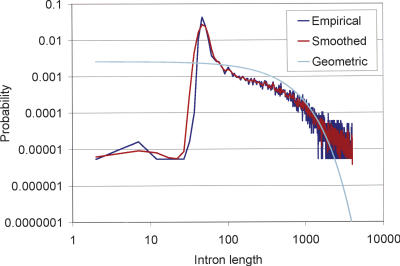
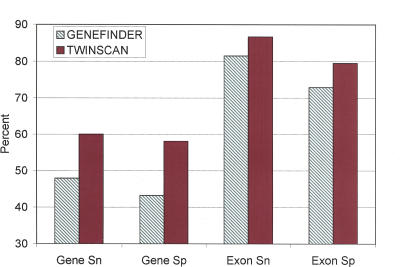
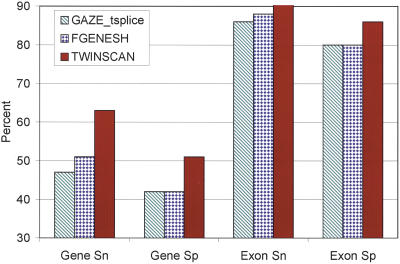
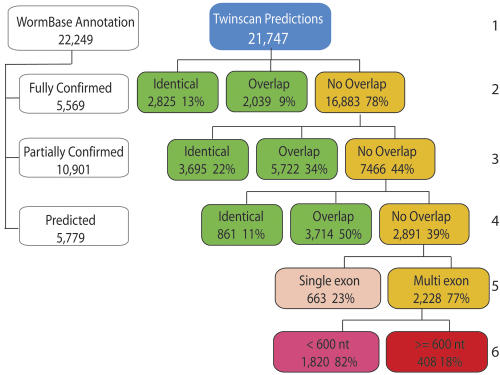
The genome of Caenorhabditis elegans was the first animal genome to be sequenced. Although considerable effort has been devoted to annotating it, the standard WormBase annotation contains thousands of predicted genes for which there is no cDNA or EST evidence. We hypothesized that a more complete experimental annotation could be obtained by creating a more accurate gene-prediction program and then amplifying and sequencing predicted genes. Our approach was to adapt the TWINSCAN gene prediction system to C. elegans and C. briggsae and to improve its splice site and intron-length models. The resulting system has 60% sensitivity and 58% specificity in exact prediction of open reading frames (ORFs), and hence, proteins-the best results we are aware of any multicellular organism. We then attempted to amplify, clone, and sequence 265 TWINSCAN-predicted ORFs that did not overlap WormBase gene annotations. The success rate was 55%, adding 146 genes that were completely absent from WormBase to the ORF clone collection (ORFeome). The same procedure had a 7% success rate on 90 Worm Base "predicted" genes that do not overlap TWINSCAN predictions. These results indicate that the accuracy of WormBase could be significantly increased by replacing its partially curated predicted genes with TWINSCAN predictions. The technology described in this study will continue to drive the C. elegans ORFeome toward completion and contribute to the annotation of the three Caenorhabditis species currently being sequenced. The results also suggest that this technology can significantly improve our knowledge of the "parts list" for even the best-studied model organisms.
Figures
Similar articles
-
C. elegans ORFeome version 3.1: increasing the coverage of ORFeome resources with improved gene predictions.Genome Res. 2004 Oct;14(10B):2064-9. doi: 10.1101/gr.2496804. Genome Res. 2004. PMID: 15489327 Free PMC article.
-
C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression.Nat Genet. 2003 May;34(1):35-41. doi: 10.1038/ng1140. Nat Genet. 2003. PMID: 12679813
-
WormBase as an integrated platform for the C. elegans ORFeome.Genome Res. 2004 Oct;14(10B):2155-61. doi: 10.1101/gr.2521304. Genome Res. 2004. PMID: 15489338 Free PMC article.
-
Comparative genomics in C. elegans, C. briggsae, and other Caenorhabditis species.Methods Mol Biol. 2006;351:13-29. doi: 10.1385/1-59745-151-7:13. Methods Mol Biol. 2006. PMID: 16988423 Review.
-
ORFeome projects: gateway between genomics and omics.Curr Opin Chem Biol. 2004 Feb;8(1):20-5. doi: 10.1016/j.cbpa.2003.12.002. Curr Opin Chem Biol. 2004. PMID: 15036152 Review.
Cited by
-
A compendium of Caenorhabditis elegans regulatory transcription factors: a resource for mapping transcription regulatory networks.Genome Biol. 2005;6(13):R110. doi: 10.1186/gb-2005-6-13-r110. Epub 2005 Dec 30. Genome Biol. 2005. PMID: 16420670 Free PMC article.
-
Massively parallel sequencing of the polyadenylated transcriptome of C. elegans.Genome Res. 2009 Apr;19(4):657-66. doi: 10.1101/gr.088112.108. Epub 2009 Jan 30. Genome Res. 2009. PMID: 19181841 Free PMC article.
-
More than 9,000,000 unique genes in human gut bacterial community: estimating gene numbers inside a human body.PLoS One. 2009 Jun 29;4(6):e6074. doi: 10.1371/journal.pone.0006074. PLoS One. 2009. PMID: 19562079 Free PMC article.
-
WormBase: better software, richer content.Nucleic Acids Res. 2006 Jan 1;34(Database issue):D475-8. doi: 10.1093/nar/gkj061. Nucleic Acids Res. 2006. PMID: 16381915 Free PMC article.
-
Genomic DNA sequence comparison between two inbred soybean cyst nematode biotypes facilitated by massively parallel 454 micro-bead sequencing.Mol Genet Genomics. 2008 May;279(5):535-43. doi: 10.1007/s00438-008-0331-8. Mol Genet Genomics. 2008. PMID: 18324416
References
-
- Brent, M.R. and Guigó, R. 2004. Recent advances in gene structure prediction. Curr. Opin. Struct. Biol. 14: 264-272. - PubMed
-
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. - PubMed
-
- C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282: 2012-2018. - PubMed
Web site references
-
- http://www.girinst.org/server/RepBase/repeatmaskerlibraries/repeatmasker...; Repeat libraries used in the foregoing analysis.
-
- http://www.sanger.ac.uk/Software/analysis/GAZE; GAZE data set.
-
- http://genes.cse.wustl.edu/eval/; Eval software.
-
- http://genes.cse.wustl.edu/wei-2005/; Predictions, primers, experimental sequences and traces, and genome alignments.
-
- http://blast.wustl.edu; Washington University BLAST archives.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials