

Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia
- PMID: 15814801
- PMCID: PMC6725382
- DOI: 10.1523/JNEUROSCI.4346-04.2005
Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia
Abstract
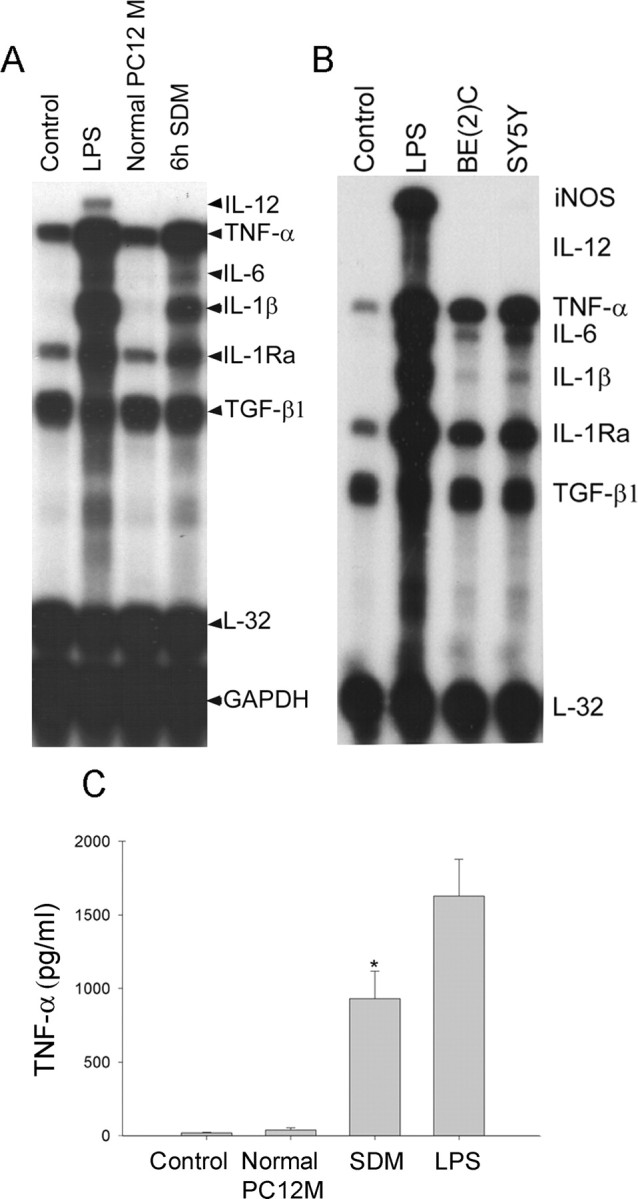
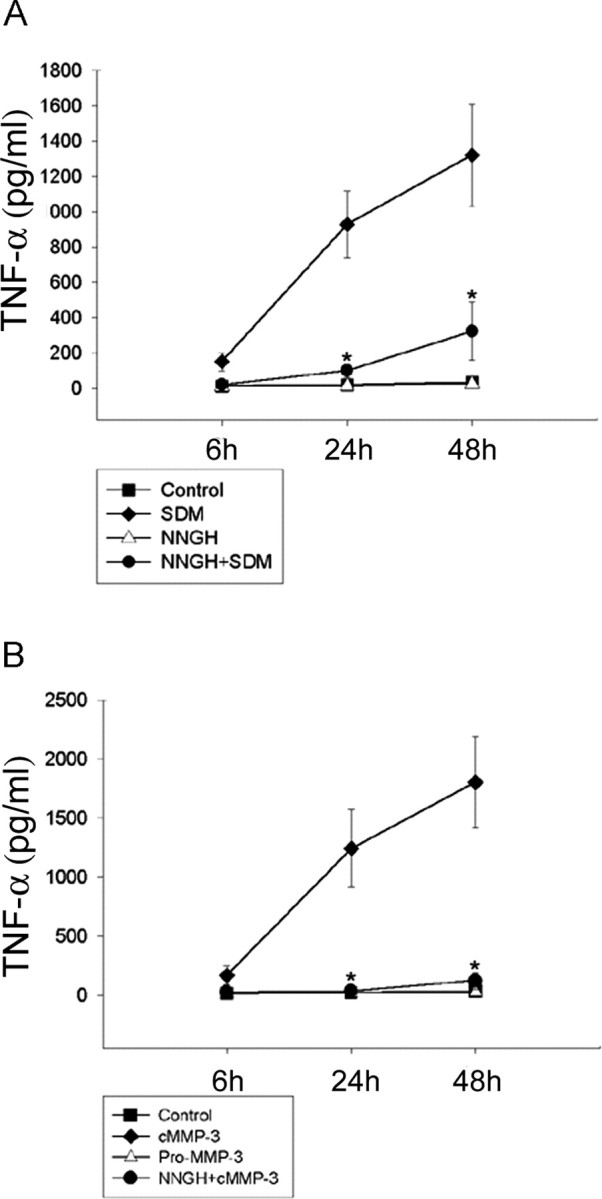
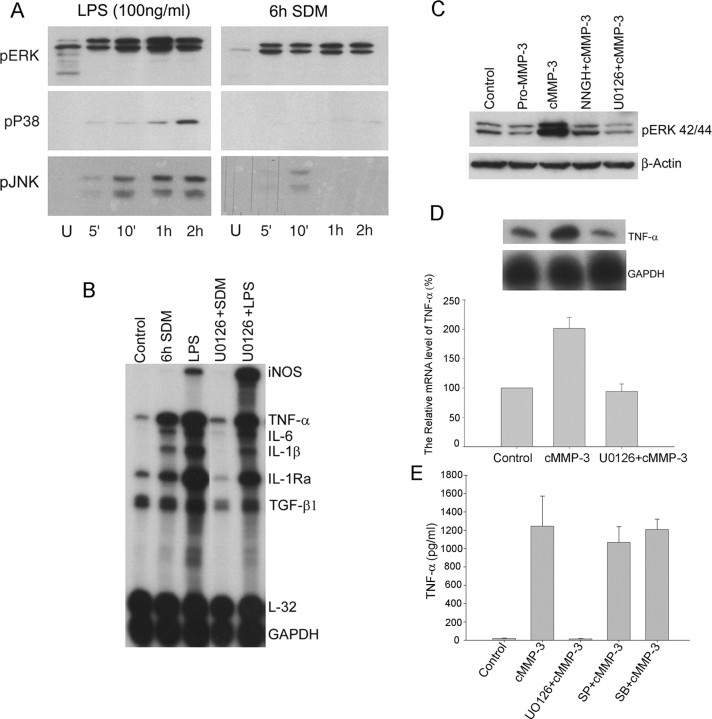
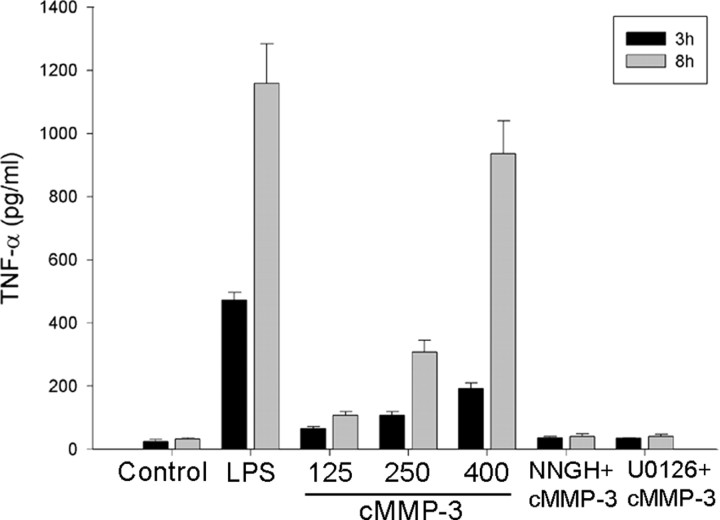
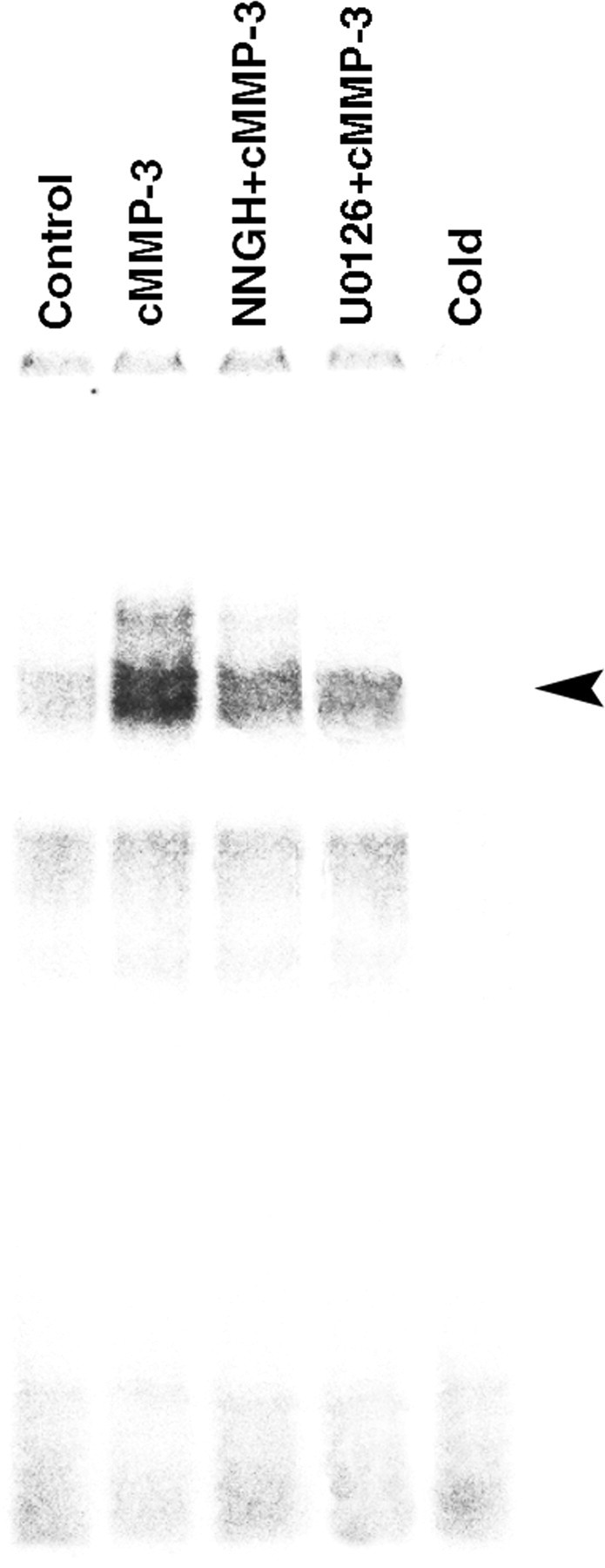
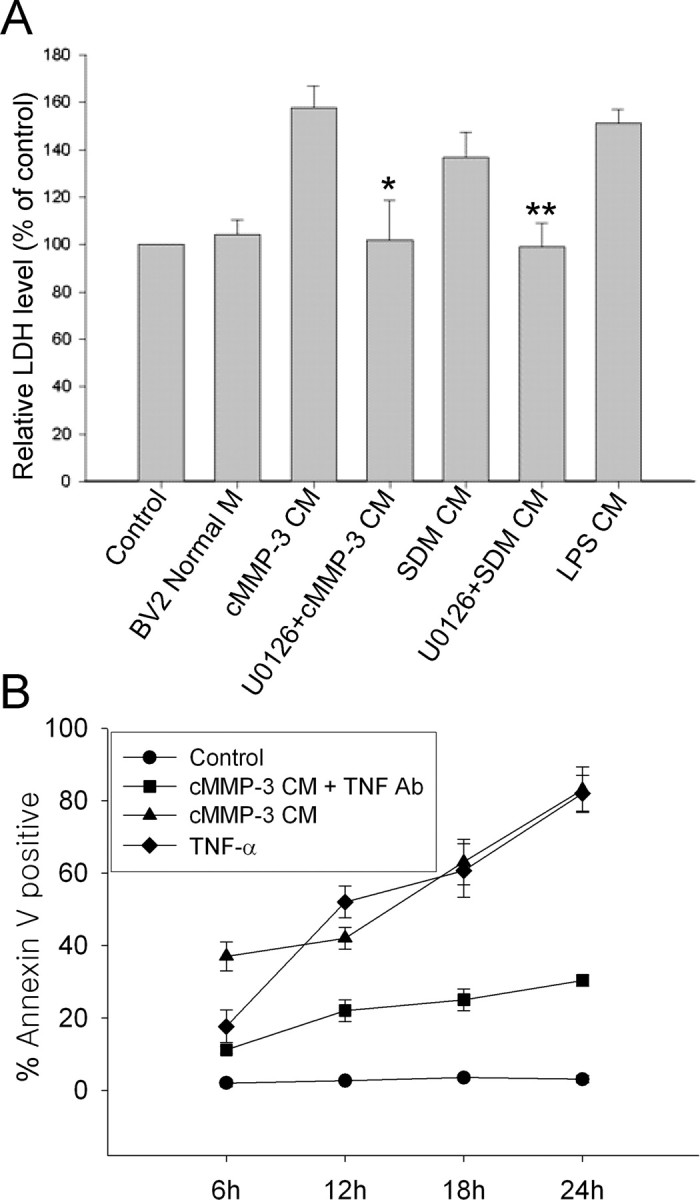
Microglial activation and inflammation are associated with progressive neuronal apoptosis in neurodegenerative human brain disorders. We sought to investigate molecular signaling mechanisms that govern activation of microglia in apoptotic neuronal degeneration. We report here that the active form of matrix metalloproteinase-3 (MMP-3) was released into the serum-deprived media (SDM) of PC12 cells and other media of apoptotic neuronal cells within 2-6 h of treatment of the cells, and SDM and catalytic domain of recombinant MMP-3 (cMMP-3) activated microglia in primary microglia cultures as well as BV2 cells, a mouse microglia cell line. Both SDM and cMMP-3 induced generation of tumor necrosis factor alpha (TNF-alpha), interleukin-6 (IL-6), IL-1beta, and interleukin-1 receptor antagonist but not IL-12 and inducible nitric oxide synthase, which are readily induced by lipopolysaccharide, in microglia, suggesting that there is a characteristic pattern of microglial cytokine induction by apoptotic neurons. Neither glial cell line-derived neurotrophic factor nor anti-inflammatory cytokines, such as IL-10 and transforming growth factor-beta1, were induced. SDM and cMMP-3 extensively released TNF-alpha from microglia and activated the nuclear factor-kappaB pathway, and these microglial responses were totally abolished by preincubation with an MMP-3 inhibitor, NNGH [N-isobutyl-N-(4-methoxyphenylsulfonyl)-glycylhydroxamic acid]. MMP-3-mediated microglial activation mostly depended on ERK (extracellular signal-regulated kinase) phosphorylation but not much on either JNK (c-Jun N-terminal protein kinase) or p38 activation. Conditioned medium of SDM- or cMMP-3-activated BV2 cells caused apoptosis of PC12 cells. These results strongly suggest that the distinctive signal of neuronal apoptosis is the release of active form of MMP-3 that activates microglia and subsequently exacerbates neuronal degeneration. Therefore, the release of MMP-3 from apoptotic neurons may play a major role in degenerative human brain disorders, such as Parkinson's disease.
Figures


Similar articles
-
Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways.Glia. 2005 Aug 1;51(2):98-110. doi: 10.1002/glia.20190. Glia. 2005. PMID: 15789435
-
Neuroprotection of Scutellarin is mediated by inhibition of microglial inflammatory activation.Neuroscience. 2011 Jun 30;185:150-60. doi: 10.1016/j.neuroscience.2011.04.005. Epub 2011 Apr 19. Neuroscience. 2011. PMID: 21524691
-
IL-1beta, an immediate early protein secreted by activated microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK and NF-kappaB pathways.J Neurosci Res. 2006 Oct;84(5):1037-46. doi: 10.1002/jnr.21011. J Neurosci Res. 2006. PMID: 16881054
-
Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease.Exp Mol Med. 2006 Aug 31;38(4):333-47. doi: 10.1038/emm.2006.40. Exp Mol Med. 2006. PMID: 16953112 Review.
-
Microglial-neuronal interactions in synaptic damage and recovery.J Neurosci Res. 1999 Oct 1;58(1):191-201. doi: 10.1002/(sici)1097-4547(19991001)58:1<191::aid-jnr17>3.0.co;2-e. J Neurosci Res. 1999. PMID: 10491582 Review.
Cited by
-
Brain inflammation and microglia: facts and misconceptions.Exp Neurobiol. 2013 Jun;22(2):59-67. doi: 10.5607/en.2013.22.2.59. Epub 2013 Jun 27. Exp Neurobiol. 2013. PMID: 23833554 Free PMC article.
-
Myelin loss associated with neuroinflammation in hypertensive rats.Stroke. 2012 Apr;43(4):1115-22. doi: 10.1161/STROKEAHA.111.643080. Epub 2012 Feb 23. Stroke. 2012. PMID: 22363061 Free PMC article.
-
The adverse effects of air pollution on the nervous system.J Toxicol. 2012;2012:782462. doi: 10.1155/2012/782462. Epub 2012 Feb 19. J Toxicol. 2012. PMID: 22523490 Free PMC article.
-
Role of oxidative stress in Parkinson's disease.Exp Neurobiol. 2013 Mar;22(1):11-7. doi: 10.5607/en.2013.22.1.11. Epub 2013 Mar 31. Exp Neurobiol. 2013. PMID: 23585717 Free PMC article.
-
Matrix metalloproteinase 3 promotes cellular anti-dengue virus response via interaction with transcription factor NFκB in cell nucleus.PLoS One. 2014 Jan 8;9(1):e84748. doi: 10.1371/journal.pone.0084748. eCollection 2014. PLoS One. 2014. PMID: 24416274 Free PMC article.
References
-
- Akassoglou K, Probert L, Kontogeorgos G, Kollias G (1997) Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol 158: 438-445. - PubMed
-
- Andersen JK (2001) Does neuronal loss in Parkinson's disease involve programmed cell death? BioEssays 23: 640-646. - PubMed
-
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW (1999) Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci 19: 1708-1716. - PMC - PubMed
-
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC (1994) Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett 172: 151-154. - PubMed
-
- Brown DR (2001) Microglia and prion disease. Microsc Res Tech 54: 71-80. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous