

Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse
- PMID: 15824086
- PMCID: PMC2213152
- DOI: 10.1084/jem.20050292
Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse
Abstract
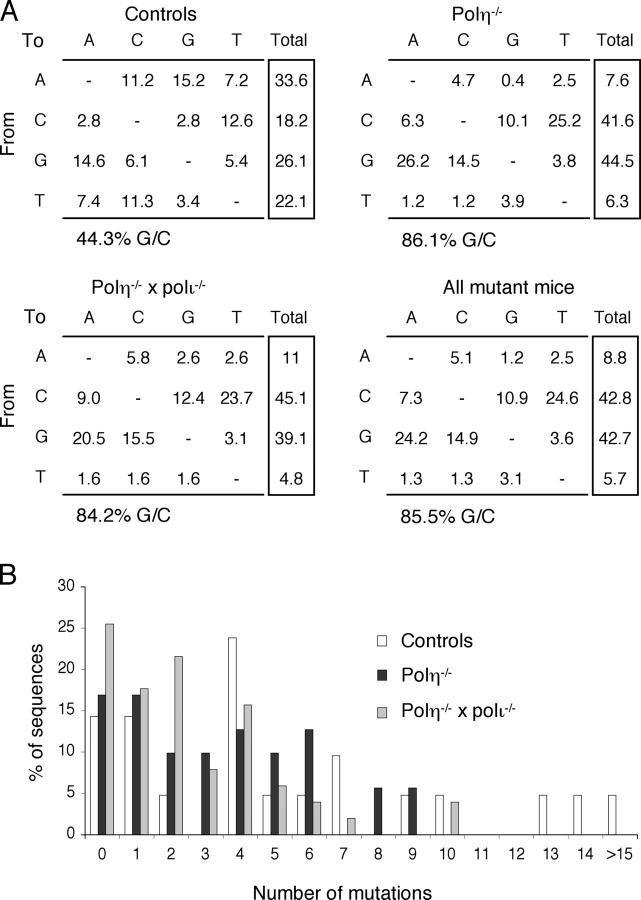
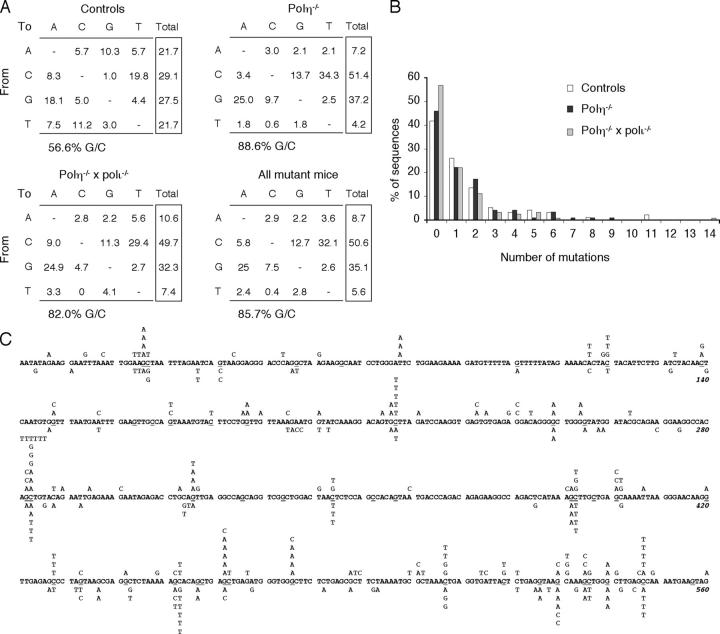
The mutation pattern of immunoglobulin genes was studied in mice deficient for DNA polymerase eta, a translesional polymerase whose inactivation is responsible for the xeroderma pigmentosum variant (XP-V) syndrome in humans. Mutations show an 85% G/C biased pattern, similar to that reported for XP-V patients. Breeding these mice with animals harboring the stop codon mutation of the 129/Olain background in their DNA polymerase iota gene did not alter this pattern further. Although this G/C biased mutation profile resembles that of mice deficient in the MSH2 or MSH6 components of the mismatch repair complex, the residual A/T mutagenesis of pol eta-deficient mice differs markedly. This suggests that, in the absence of pol eta, the MSH2-MSH6 complex is able to recruit another DNA polymerase that is more accurate at copying A/T bases, possibly pol kappa, to assume its function in hypermutation.
Figures
References
-
- Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. - PubMed
-
- Barreto, V.M., A.R. Ramiro, and M.C. Nussenzweig. 2005. Activation-induced deaminase: controversies and open questions. Trends Immunol. 26:90–96. - PubMed
-
- Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. - PubMed
-
- Reynaud, C.-A., S. Aoufouchi, A. Faili, and J.-C. Weill. 2003. What role for AID: mutator, or assembler of the immunoglobulin mutasome? Nat. Immunol. 4:631–638. - PubMed
-
- Rada, C., J.M. Di Noia, and M.S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T focused phase of somatic mutation. Mol. Cell. 16:163–171. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
