

An efficient Monte Carlo method for estimating Ne from temporally spaced samples using a coalescent-based likelihood
- PMID: 15834143
- PMCID: PMC1450415
- DOI: 10.1534/genetics.104.038349
An efficient Monte Carlo method for estimating Ne from temporally spaced samples using a coalescent-based likelihood
Abstract
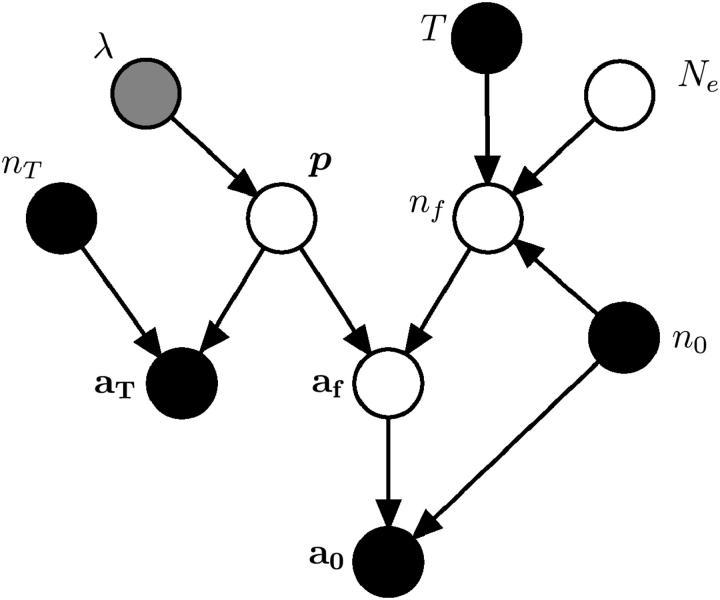
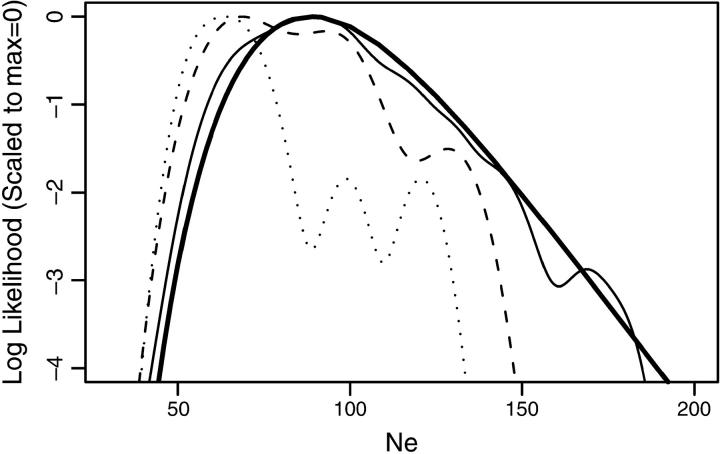
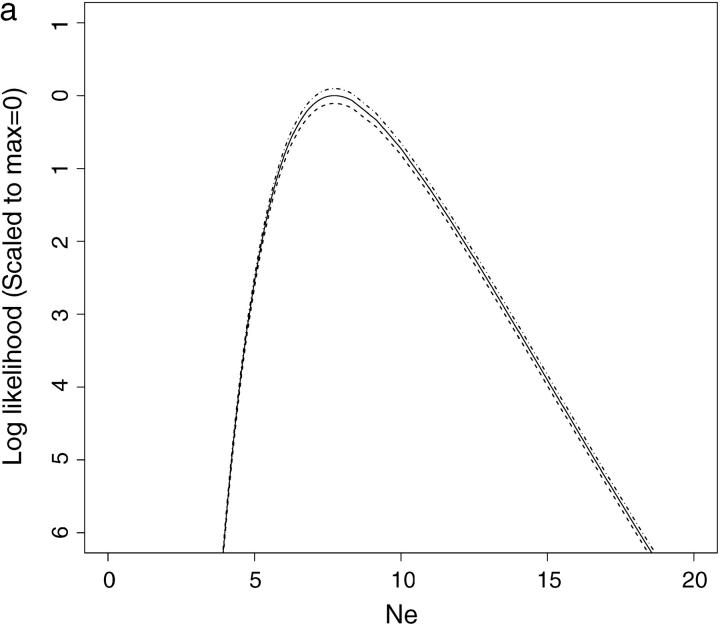
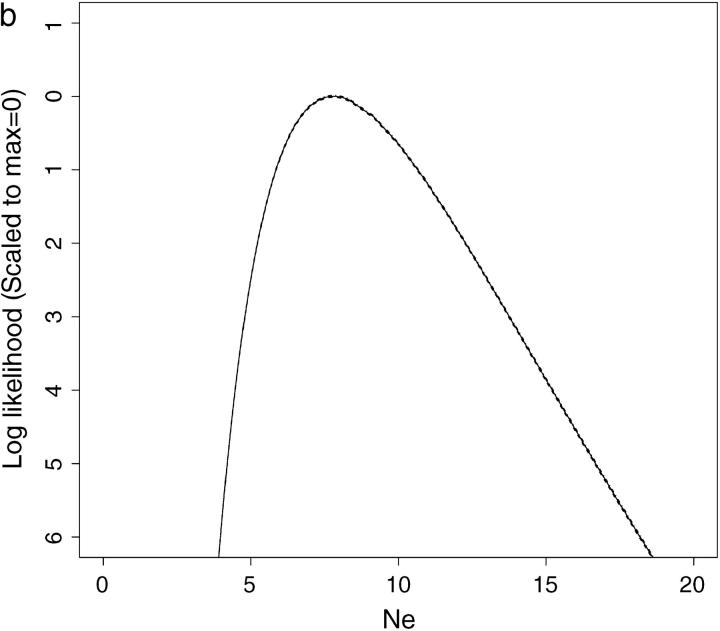
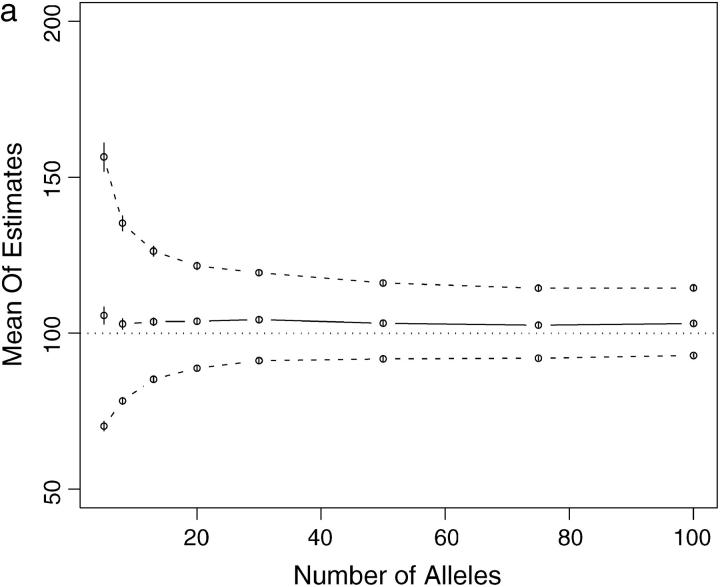
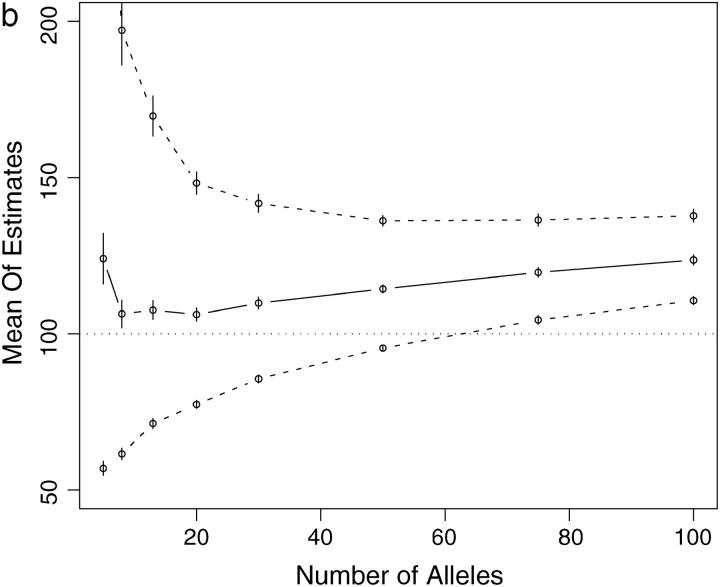
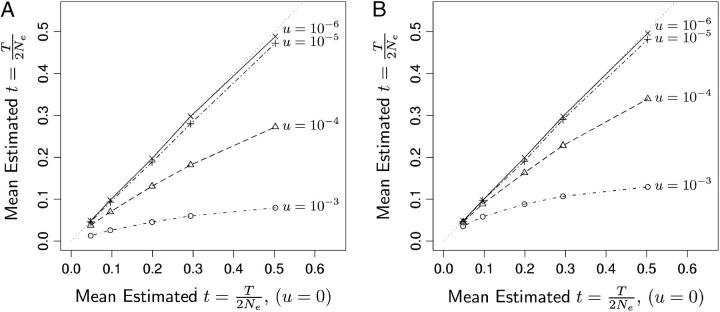
This article presents an efficient importance-sampling method for computing the likelihood of the effective size of a population under the coalescent model of Berthier et al. Previous computational approaches, using Markov chain Monte Carlo, required many minutes to several hours to analyze small data sets. The approach presented here is orders of magnitude faster and can provide an approximation to the likelihood curve, even for large data sets, in a matter of seconds. Additionally, confidence intervals on the estimated likelihood curve provide a useful estimate of the Monte Carlo error. Simulations show the importance sampling to be stable across a wide range of scenarios and show that the N(e) estimator itself performs well. Further simulations show that the 95% confidence intervals around the N(e) estimate are accurate. User-friendly software implementing the algorithm for Mac, Windows, and Unix/Linux is available for download. Applications of this computational framework to other problems are discussed.
Figures
References
-
- Baum, L. E., 1971. Statistical inference for probabilistic functions of finite state Markov chains. Ann. Math. Stat. 37: 1554–1563.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
