

Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN
- PMID: 15837813
- PMCID: PMC2213143
- DOI: 10.1084/jem.20041276
Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN
Abstract
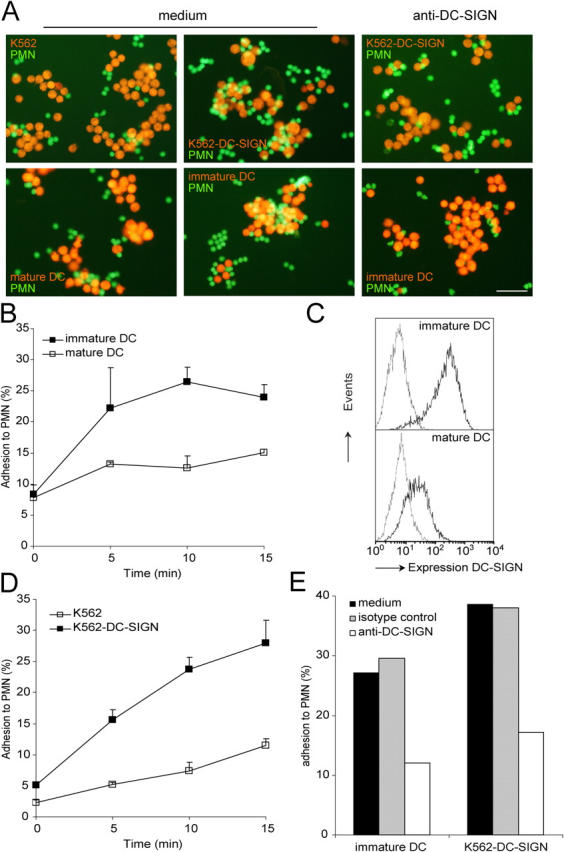
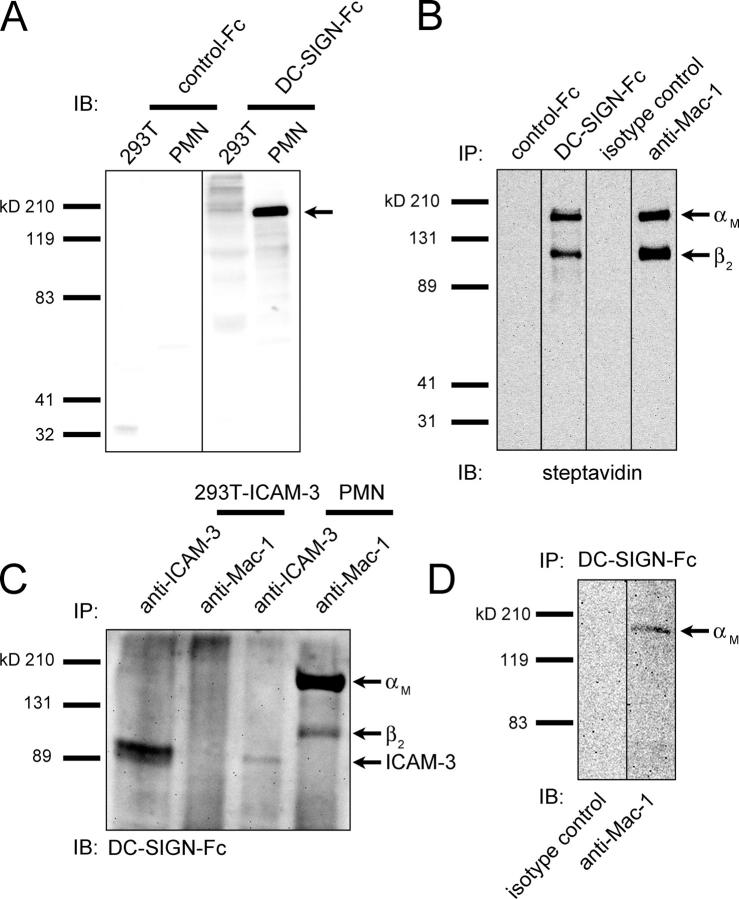
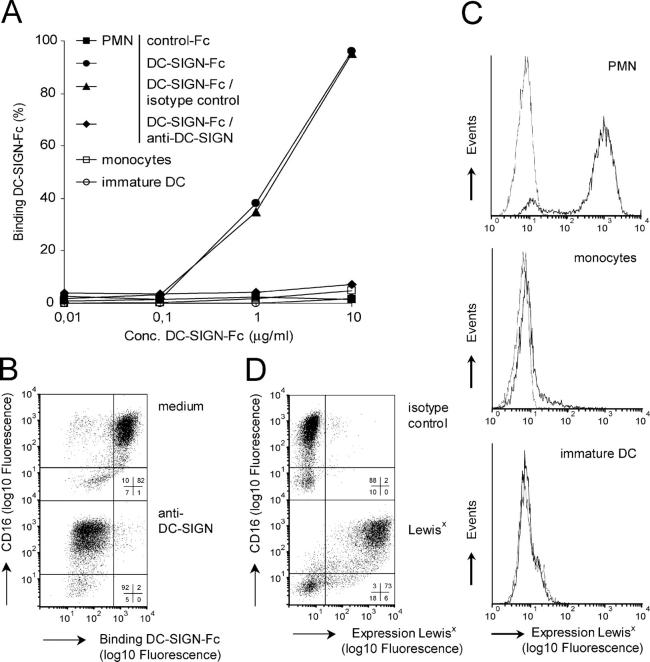
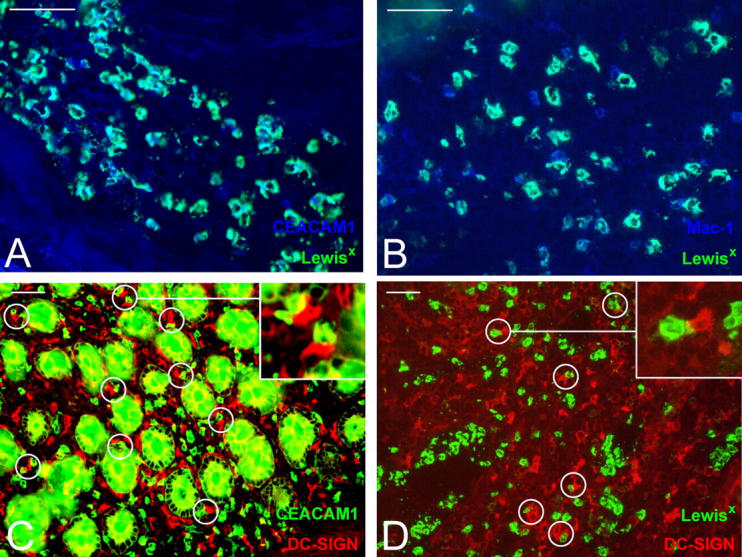
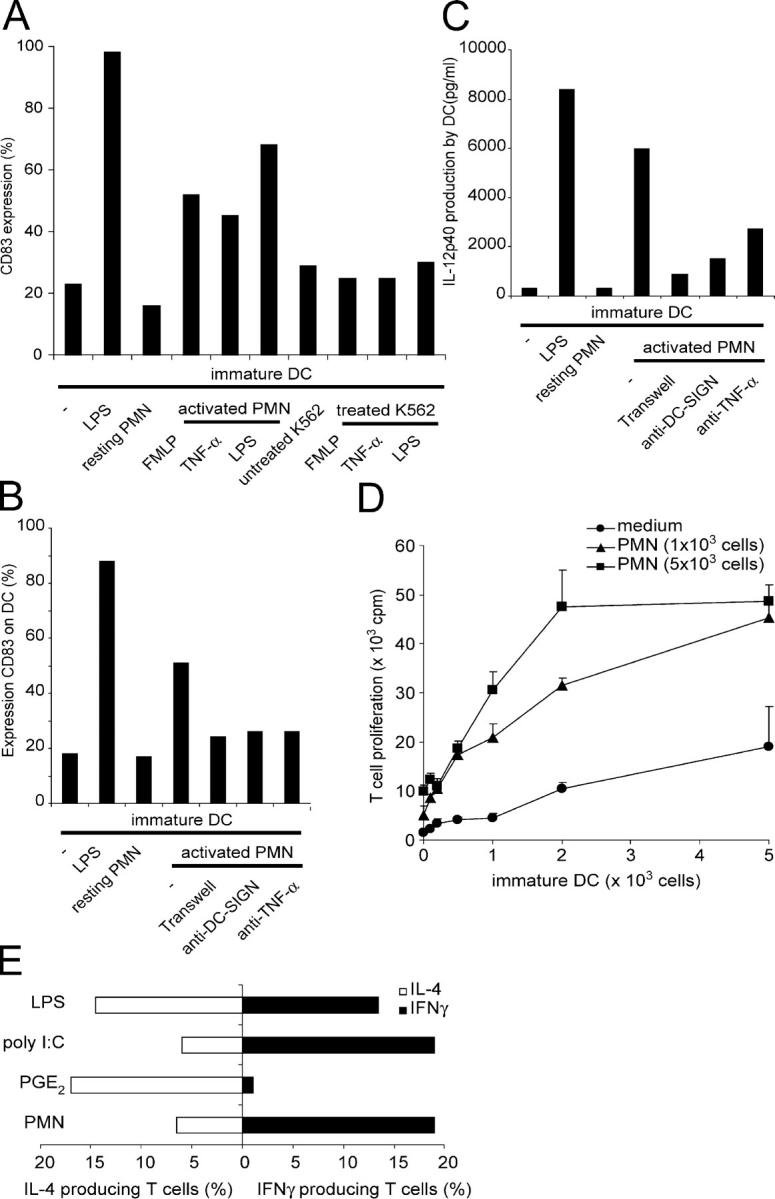
Neutrophils are key players of the innate immune system that provide a first line of defense against invading pathogens. However, it is unknown whether neutrophils can interact with dendritic cells (DCs) to modulate adaptive immune responses. We demonstrate that neutrophils strongly cluster with immature DCs and that activated, not resting, neutrophils induce maturation of DCs that enables these DCs to trigger strong T cell proliferation and T helper type 1 polarization of T cells. This neutrophil-DC interaction is driven by the binding of the DC-specific, C-type lectin DC-SIGN to the beta(2)-integrin Mac-1. Strikingly, DC-SIGN only interacts with Mac-1 from neutrophils, but not from other leukocytes, mainly because of specific Lewis(x) carbohydrates that are present on the alpha(M) chain of Mac-1 from neutrophils. Furthermore, we show that besides the formation of cellular contact, the tumor necrosis factor-alpha produced by activated neutrophils is essential for inducing DC maturation. Our data demonstrate that DC-SIGN and Mac-1 define a molecular pathway to establish cellular adhesion between DCs and neutrophils, thereby providing a novel cellular link between innate and adaptive immunity.
Figures


References
-
- Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. - PubMed
-
- Geijtenbeek, T.B.H., R. Torensma, S.J. van Vliet, G.C.F. van Duijnhoven, G.J. Adema, Y. van Kooyk, and C.G. Figdor. 2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 100:575–585. - PubMed
-
- Geijtenbeek, T.B., D.S. Kwon, R. Torensma, S.J. van Vliet, G.C.F. van Duijnhoven, J. Middel, I.L.M.H. Cornelissen, H.S. Nottet, V.N. KewalRamani, D.R. Littman, C.G. Figdor, and Y. van Kooyk. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 100:587–597. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
