

All are not equal: a benchmark of different homology modeling programs
- PMID: 15840834
- PMCID: PMC2253266
- DOI: 10.1110/ps.041253405
All are not equal: a benchmark of different homology modeling programs
Abstract
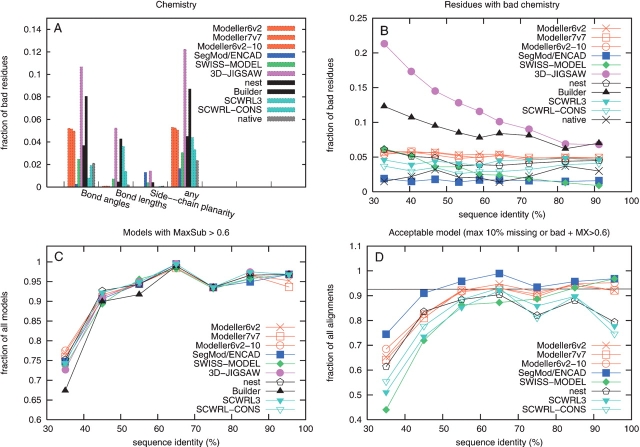
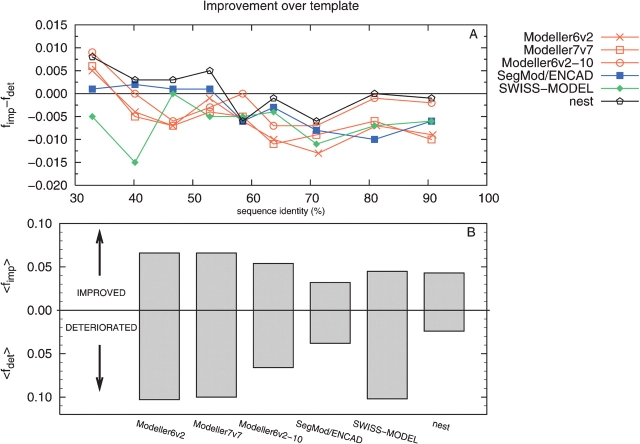
Modeling a protein structure based on a homologous structure is a standard method in structural biology today. In this process an alignment of a target protein sequence onto the structure of a template(s) is used as input to a program that constructs a 3D model. It has been shown that the most important factor in this process is the correctness of the alignment and the choice of the best template structure(s), while it is generally believed that there are no major differences between the best modeling programs. Therefore, a large number of studies to benchmark the alignment qualities and the selection process have been performed. However, to our knowledge no large-scale benchmark has been performed to evaluate the programs used to transform the alignment to a 3D model. In this study, a benchmark of six different homology modeling programs- Modeller, SegMod/ENCAD, SWISS-MODEL, 3D-JIGSAW, nest, and Builder-is presented. The performance of these programs is evaluated using physiochemical correctness and structural similarity to the correct structure. From our analysis it can be concluded that no single modeling program outperform the others in all tests. However, it is quite clear that three modeling programs, Modeller, nest, and SegMod/ ENCAD, perform better than the others. Interestingly, the fastest and oldest modeling program, SegMod/ ENCAD, performs very well, although it was written more than 10 years ago and has not undergone any development since. It can also be observed that none of the homology modeling programs builds side chains as well as a specialized program (SCWRL), and therefore there should be room for improvement.
Figures




References
-
- Abagyan, R.A. and Batalov, S. 1997. Do aligned sequences share the same fold? J. Mol. Biol. 273 355–368. - PubMed
-
- Bates, P.A., Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2001. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins 45 (Suppl. 5): 39–46. - PubMed
-
- Blundell, T.L., Sibanda, B.L., Sternberg, M.J., and Thornton, J.M. 1987. Knowledge-based prediction of protein structures and the design of novel molecules. Nature 326 347–352. - PubMed
-
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4 187–217.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
