

Synergy between a plasminogen cascade and MMP-9 in autoimmune disease
- PMID: 15841177
- PMCID: PMC1070424
- DOI: 10.1172/JCI23977
Synergy between a plasminogen cascade and MMP-9 in autoimmune disease
Abstract
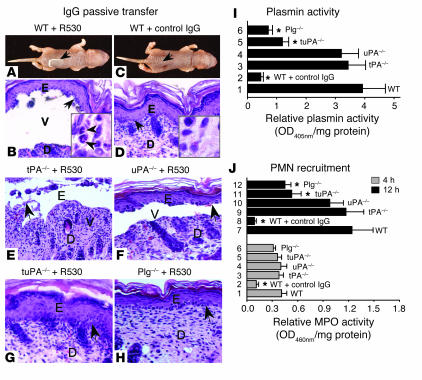
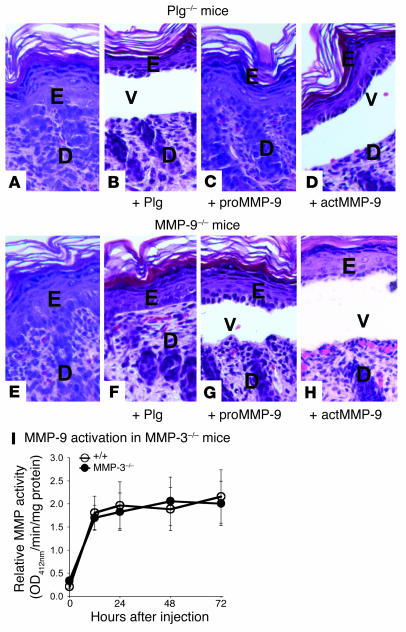
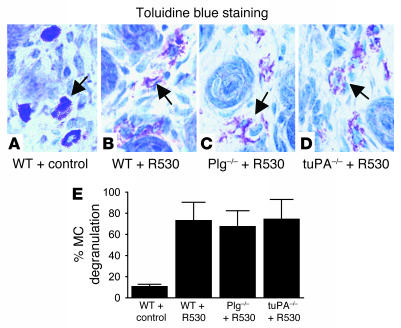
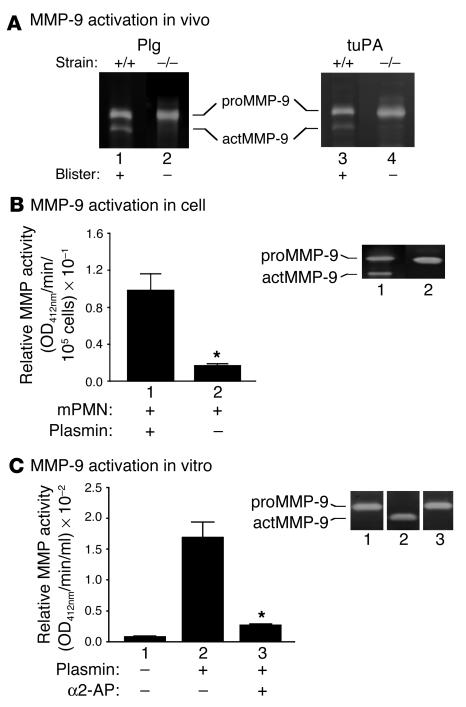
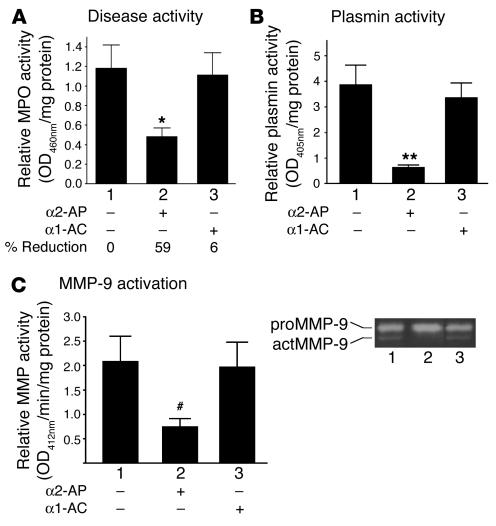
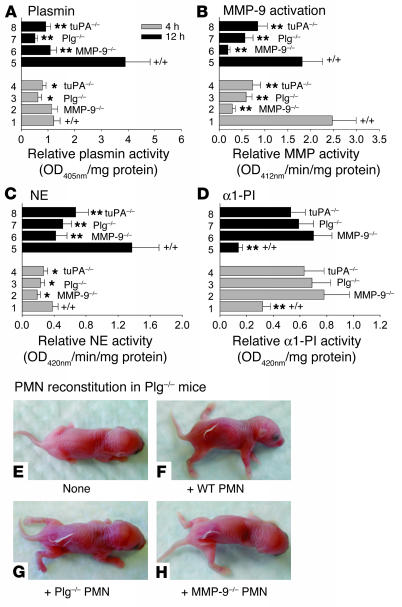
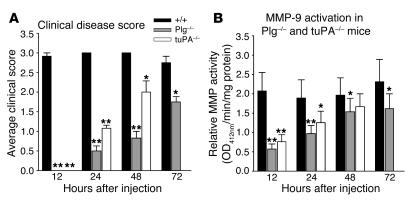
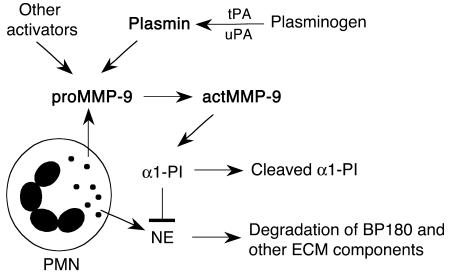
Extracellular proteolysis by the plasminogen/plasmin (Plg/plasmin) system and MMPs is required for tissue injury in autoimmune and inflammatory diseases. We demonstrate that a Plg cascade synergizes with MMP-9/gelatinase B in vivo during dermal-epidermal separation in an experimental model of bullous pemphigoid (BP), an autoimmune disease. BP was induced in mice by antibodies to the hemidesmosomal antigen BP180. Mice deficient in MMP-9 were resistant to experimental BP, while mice deficient in Plg and both tissue Plg activator (tPA) and urokinase Plg activator (uPA) showed delayed and less intense blister formation induced by antibodies to BP180. Plg-deficient mice reconstituted locally with Plg or the active form of MMP-9 (actMMP-9), but not the proenzyme form of MMP-9 (proMMP-9), developed BP. In contrast, proMMP-9 or actMMP-9, but not Plg, reconstituted susceptibility of MMP-9-deficient mice to the skin disease. In addition, MMP-3-deficient mice injected with pathogenic IgG developed the same degree of BP and expressed levels of actMMP-9 in the skin similar to those of WT controls. Thus, the Plg/plasmin system is epistatic to MMP-9 activation and subsequent dermal-epidermal separation in BP.
Figures
Comment in
-
The pathophysiology of autoimmune blistering diseases.J Clin Invest. 2005 Apr;115(4):825-8. doi: 10.1172/JCI24855. J Clin Invest. 2005. PMID: 15841169 Free PMC article.
References
-
- Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. - PubMed
-
- Collen D. The plasminogen (fibrinolytic) system. Thromb. Haemost. 1999;82:259–270. - PubMed
-
- Werb Z, Mainardi CL, Vater CA, Harris ED., Jr Endogenous activation of latent collagenase by rheumatoid synovial cells. Evidence for a role for plasminogen activator. N. Engl. J. Med. 1977;296:1017–1023. - PubMed
-
- Woessner, J.F., and Nagase, H. 2000. Matrix metalloproteinases and TIMPs. Oxford University Press. Oxford, United Kingdom. 240 pp.
-
- Stanley, J.R. 1999. Bullous pemphigoid. In Fitzpatrick’s dermatology in general medicine. I.M. Freedberg et al., editors. McGraw-Hill. New York, New York, USA. 666–671.
Publication types
MeSH terms
Substances
Grants and funding
- R01 AI061430/AI/NIAID NIH HHS/United States
- R29 AI040768/AI/NIAID NIH HHS/United States
- P01 AI053194/AI/NIAID NIH HHS/United States
- R01 AI040768/AI/NIAID NIH HHS/United States
- R37 AR032081/AR/NIAMS NIH HHS/United States
- CA72006/CA/NCI NIH HHS/United States
- AR32599/AR/NIAMS NIH HHS/United States
- R01 AR032081/AR/NIAMS NIH HHS/United States
- R01 HL047328/HL/NHLBI NIH HHS/United States
- AI40768/AI/NIAID NIH HHS/United States
- AI53194/AI/NIAID NIH HHS/United States
- R01 AR032599/AR/NIAMS NIH HHS/United States
- P01 CA072006/CA/NCI NIH HHS/United States
- HL47328/HL/NHLBI NIH HHS/United States
- AR32081/AR/NIAMS NIH HHS/United States
- AI61430/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
