

The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism
- PMID: 15841214
- PMCID: PMC1077171
- DOI: 10.1172/JCI22782
The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism
Abstract
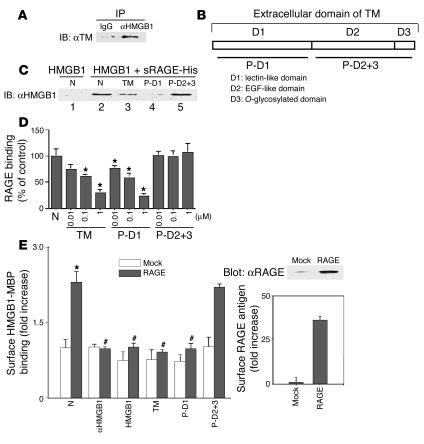
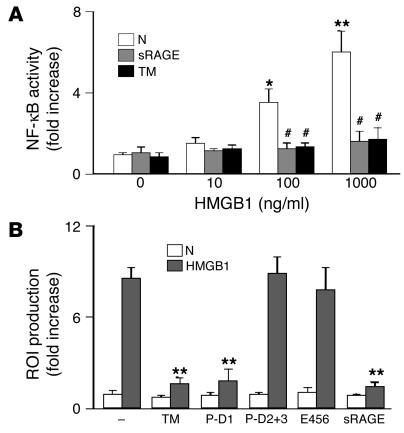
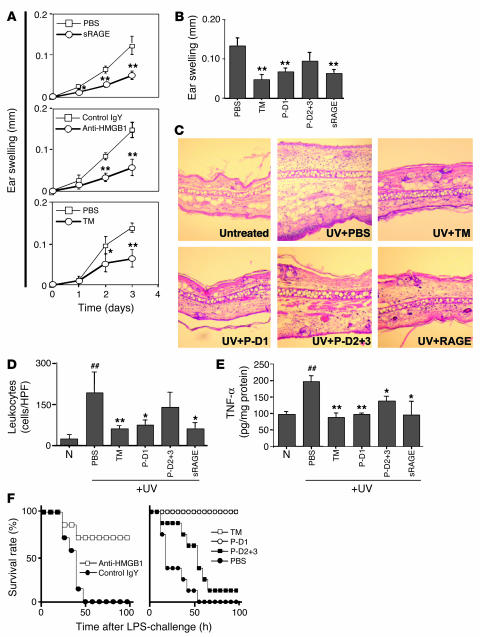
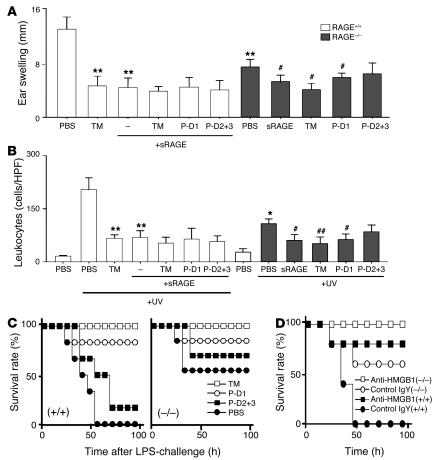
Thrombomodulin (TM) is an endothelial anticoagulant cofactor that promotes thrombin-mediated formation of activated protein C (APC). We have found that the N-terminal lectin-like domain (D1) of TM has unique antiinflammatory properties. TM, via D1, binds high-mobility group-B1 DNA-binding protein (HMGB1), a factor closely associated with necrotic cell damage following its release from the nucleus, thereby preventing in vitro leukocyte activation, in vivo UV irradiation-induced cutaneous inflammation, and in vivo lipopolysaccharide-induced lethality. Our data also demonstrate antiinflammatory properties of a peptide spanning D1 of TM and suggest its therapeutic potential. These findings highlight a novel mechanism, i.e., sequestration of mediators, through which an endothelial cofactor, TM, suppresses inflammation quite distinctly from its anticoagulant cofactor activity, thereby preventing the interaction of these mediators with cell surface receptors on effector cells in the vasculature.
Figures
References
-
- Esmon CT, Esmon NL, Harris KW. Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor V activation. J. Biol. Chem. 1982;257:7944–7947. - PubMed
-
- Esmon CT. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem. 1989;264:4743–4746. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
