

Scoredist: a simple and robust protein sequence distance estimator
- PMID: 15857510
- PMCID: PMC1131889
- DOI: 10.1186/1471-2105-6-108
Scoredist: a simple and robust protein sequence distance estimator
Abstract
Background: Distance-based methods are popular for reconstructing evolutionary trees thanks to their speed and generality. A number of methods exist for estimating distances from sequence alignments, which often involves some sort of correction for multiple substitutions. The problem is to accurately estimate the number of true substitutions given an observed alignment. So far, the most accurate protein distance estimators have looked for the optimal matrix in a series of transition probability matrices, e.g. the Dayhoff series. The evolutionary distance between two aligned sequences is here estimated as the evolutionary distance of the optimal matrix. The optimal matrix can be found either by an iterative search for the Maximum Likelihood matrix, or by integration to find the Expected Distance. As a consequence, these methods are more complex to implement and computationally heavier than correction-based methods. Another problem is that the result may vary substantially depending on the evolutionary model used for the matrices. An ideal distance estimator should produce consistent and accurate distances independent of the evolutionary model used.
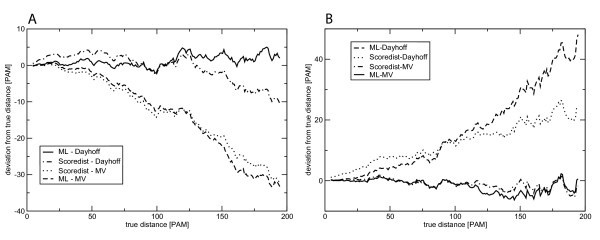
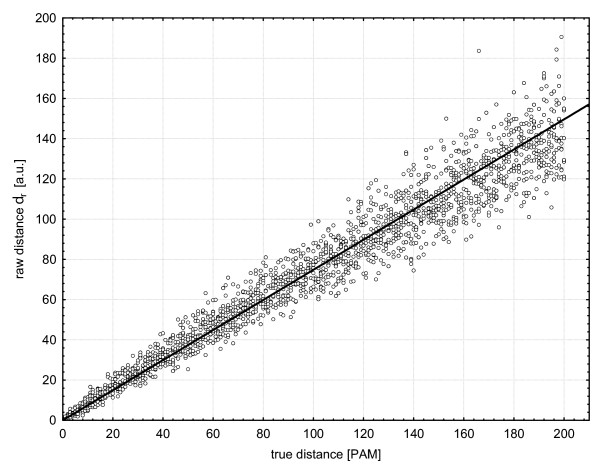
Results: We propose a correction-based protein sequence estimator called Scoredist. It uses a logarithmic correction of observed divergence based on the alignment score according to the BLOSUM62 score matrix. We evaluated Scoredist and a number of optimal matrix methods using three evolutionary models for both training and testing Dayhoff, Jones-Taylor-Thornton, and Muller-Vingron, as well as Whelan and Goldman solely for testing. Test alignments with known distances between 0.01 and 2 substitutions per position (1-200 PAM) were simulated using ROSE. Scoredist proved as accurate as the optimal matrix methods, yet substantially more robust. When trained on one model but tested on another one, Scoredist was nearly always more accurate. The Jukes-Cantor and Kimura correction methods were also tested, but were substantially less accurate.
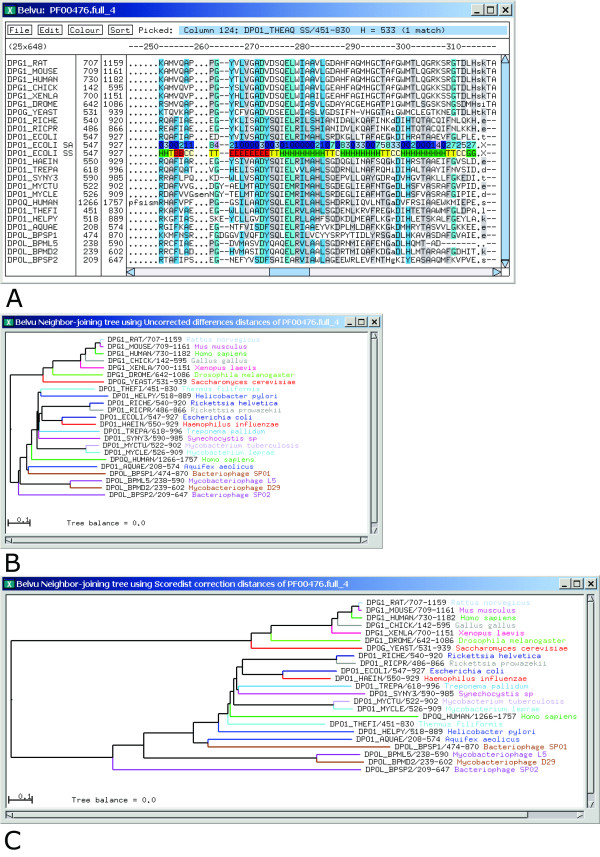
Conclusion: The Scoredist distance estimator is fast to implement and run, and combines robustness with accuracy. Scoredist has been incorporated into the Belvu alignment viewer, which is available at ftp://ftp.cgb.ki.se/pub/prog/belvu/.
Figures
Similar articles
-
Assessment of protein distance measures and tree-building methods for phylogenetic tree reconstruction.Mol Biol Evol. 2005 Nov;22(11):2257-64. doi: 10.1093/molbev/msi224. Epub 2005 Jul 27. Mol Biol Evol. 2005. PMID: 16049194
-
On the quality of tree-based protein classification.Bioinformatics. 2005 May 1;21(9):1876-90. doi: 10.1093/bioinformatics/bti244. Epub 2005 Jan 12. Bioinformatics. 2005. PMID: 15647305
-
Toward extracting all phylogenetic information from matrices of evolutionary distances.Science. 2010 Mar 12;327(5971):1376-9. doi: 10.1126/science.1182300. Science. 2010. PMID: 20223986
-
Testing substitution models within a phylogenetic tree.Mol Biol Evol. 2003 Apr;20(4):572-8. doi: 10.1093/molbev/msg073. Epub 2003 Apr 2. Mol Biol Evol. 2003. PMID: 12679552 Review.
-
Where did the BLOSUM62 alignment score matrix come from?Nat Biotechnol. 2004 Aug;22(8):1035-6. doi: 10.1038/nbt0804-1035. Nat Biotechnol. 2004. PMID: 15286655 Review.
Cited by
-
Towards a practical O(nlogn) phylogeny algorithm.Algorithms Mol Biol. 2012 Nov 26;7(1):32. doi: 10.1186/1748-7188-7-32. Algorithms Mol Biol. 2012. PMID: 23181935 Free PMC article.
-
Adaptive evolution has targeted the C-terminal domain of the RXLR effectors of plant pathogenic oomycetes.Plant Cell. 2007 Aug;19(8):2349-69. doi: 10.1105/tpc.107.051037. Epub 2007 Aug 3. Plant Cell. 2007. PMID: 17675403 Free PMC article.
-
Isopentenyltransferase-1 (IPT1) knockout in Physcomitrella together with phylogenetic analyses of IPTs provide insights into evolution of plant cytokinin biosynthesis.J Exp Bot. 2014 Jun;65(9):2533-43. doi: 10.1093/jxb/eru142. Epub 2014 Apr 1. J Exp Bot. 2014. PMID: 24692654 Free PMC article.
-
The evolution of nuclear auxin signalling.BMC Evol Biol. 2009 Jun 3;9:126. doi: 10.1186/1471-2148-9-126. BMC Evol Biol. 2009. PMID: 19493348 Free PMC article.
-
Analysis of genome sequences from plant pathogenic Rhodococcus reveals genetic novelties in virulence loci.PLoS One. 2014 Jul 10;9(7):e101996. doi: 10.1371/journal.pone.0101996. eCollection 2014. PLoS One. 2014. PMID: 25010934 Free PMC article.
References
-
- Bruno WJ, Socci ND, Halpern AL. Weighted Neighbor Joining: A Likelihood-Based Approach to Distance-Based Phylogeny Reconstruction. Mol Biol Evol. 2000;17:189–197. - PubMed
-
- Gascuel O. BIONJ: An Improved Version on the NJ Algorithm Based on a Simple Model of Sequence Data. Mol Biol Evol. 1997;14:685–695. - PubMed
-
- Saitou N, Nei M. The Neighbor-joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol Biol Evol. 1987;4:406–425. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
