

A benchmark of multiple sequence alignment programs upon structural RNAs
- PMID: 15860779
- PMCID: PMC1087786
- DOI: 10.1093/nar/gki541
A benchmark of multiple sequence alignment programs upon structural RNAs
Abstract
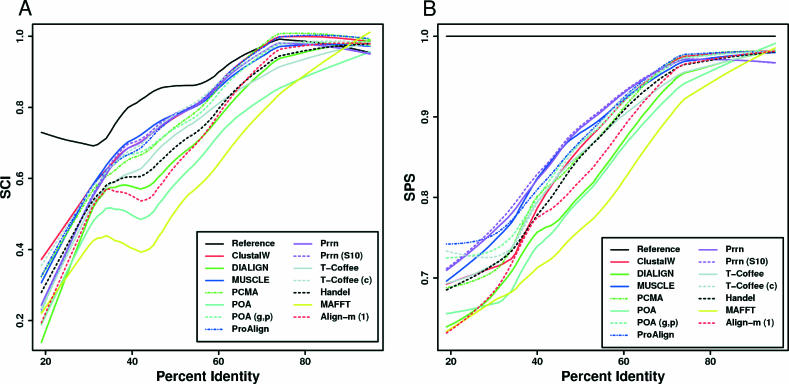
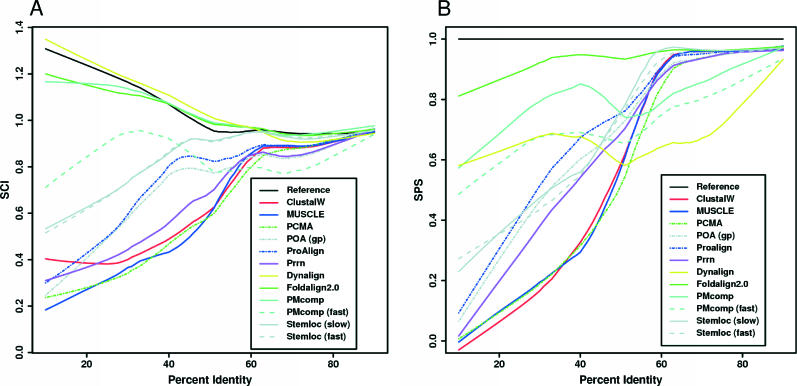
To date, few attempts have been made to benchmark the alignment algorithms upon nucleic acid sequences. Frequently, sophisticated PAM or BLOSUM like models are used to align proteins, yet equivalents are not considered for nucleic acids; instead, rather ad hoc models are generally favoured. Here, we systematically test the performance of existing alignment algorithms on structural RNAs. This work was aimed at achieving the following goals: (i) to determine conditions where it is appropriate to apply common sequence alignment methods to the structural RNA alignment problem. This indicates where and when researchers should consider augmenting the alignment process with auxiliary information, such as secondary structure and (ii) to determine which sequence alignment algorithms perform well under the broadest range of conditions. We find that sequence alignment alone, using the current algorithms, is generally inappropriate <50-60% sequence identity. Second, we note that the probabilistic method ProAlign and the aging Clustal algorithms generally outperform other sequence-based algorithms, under the broadest range of applications.
Figures
References
-
- Chiu D.K., Kolodziejczak T. Inferring consensus structure from nucleic acid sequences. Comput. Appl. Biosci. 1991;7:347–352. - PubMed
-
- Gorodkin J., Heyer L., Brunak S., Stormo G. Displaying the information contents of structural RNA alignments. CABIOS. 1997;13:583–586. - PubMed
-
- Hofacker I., Fekete M., Stadler P. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
