

Mechanically unfolding the small, topologically simple protein L
- PMID: 15863479
- PMCID: PMC1366550
- DOI: 10.1529/biophysj.105.061465
Mechanically unfolding the small, topologically simple protein L
Abstract
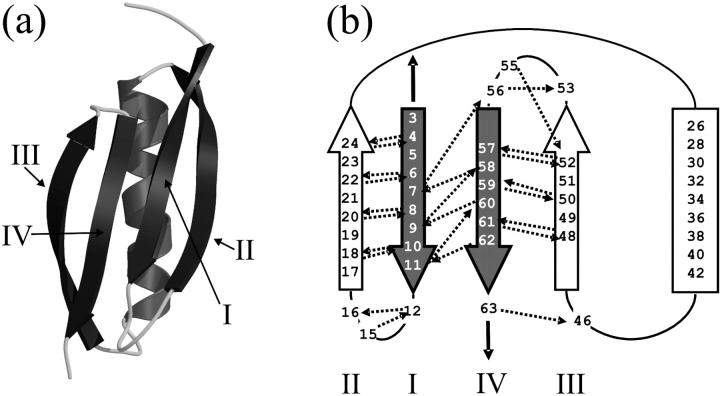
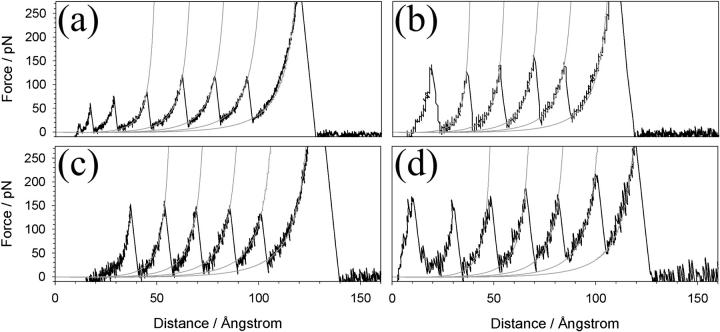
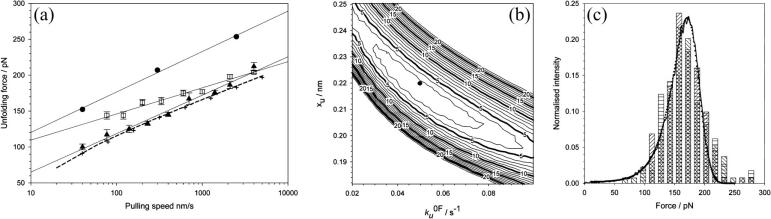
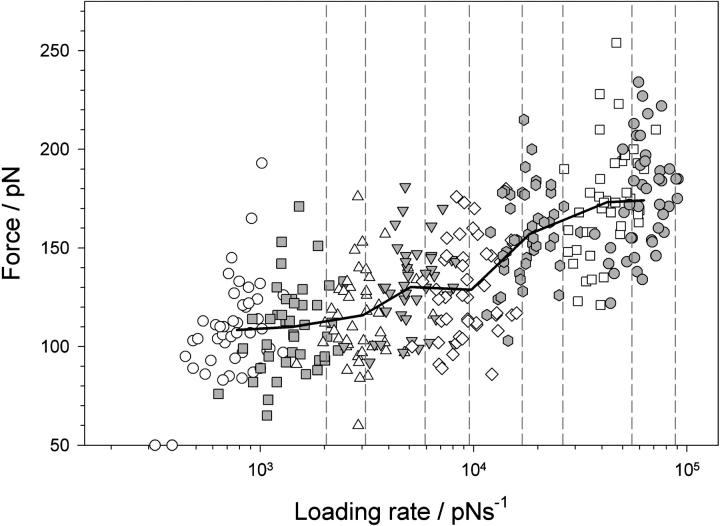
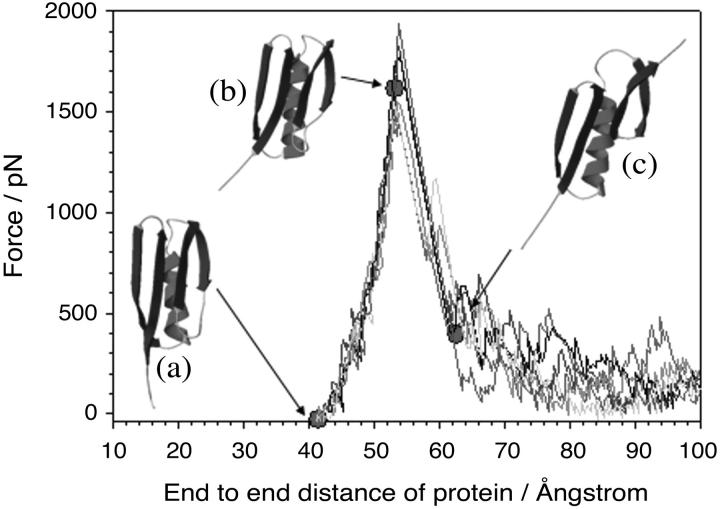
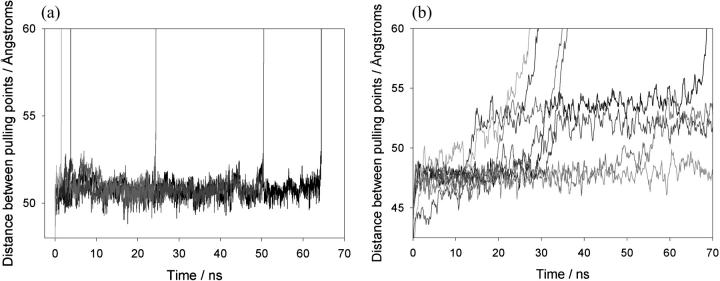
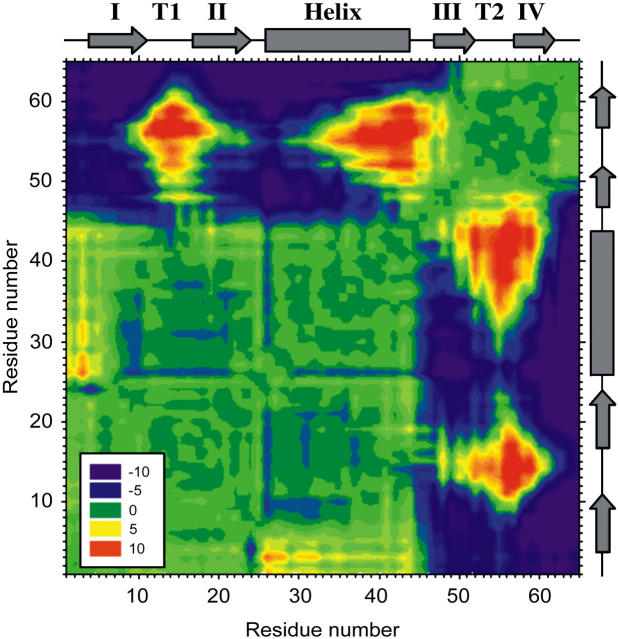
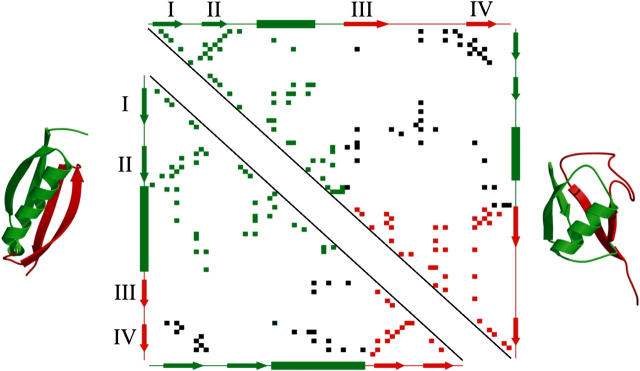
beta-sheet proteins are generally more able to resist mechanical deformation than alpha-helical proteins. Experiments measuring the mechanical resistance of beta-sheet proteins extended by their termini led to the hypothesis that parallel, directly hydrogen-bonded terminal beta-strands provide the greatest mechanical strength. Here we test this hypothesis by measuring the mechanical properties of protein L, a domain with a topology predicted to be mechanically strong, but with no known mechanical function. A pentamer of this small, topologically simple protein is resistant to mechanical deformation over a wide range of extension rates. Molecular dynamics simulations show the energy landscape for protein L is highly restricted for mechanical unfolding and that this protein unfolds by the shearing apart of two structural units in a mechanism similar to that proposed for ubiquitin, which belongs to the same structural class as protein L, but unfolds at a significantly higher force. These data suggest that the mechanism of mechanical unfolding is conserved in proteins within the same fold family and demonstrate that although the topology and presence of a hydrogen-bonded clamp are of central importance in determining mechanical strength, hydrophobic interactions also play an important role in modulating the mechanical resistance of these similar proteins.
Figures
References
-
- Mitsui, K., M. Hara, and A. Ikai. 1996. Mechanical unfolding of α(2)-macroglobulin molecules with atomic force microscope. FEBS Lett. 385:29–33. - PubMed
-
- Carrion-Vazquez, M., H. B. Li, H. Lu, P. E. Marszalek, A. F. Oberhauser, and J. M. Fernandez. 2003. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol. 10:738–743. - PubMed
-
- Rief, M., J. Pascual, M. Saraste, and H. E. Gaub. 1999. Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles. J. Mol. Biol. 286:553–561. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
