

hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair
- PMID: 15864295
- PMCID: PMC1299302
- DOI: 10.1038/sj.embor.7400392
hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair
Abstract
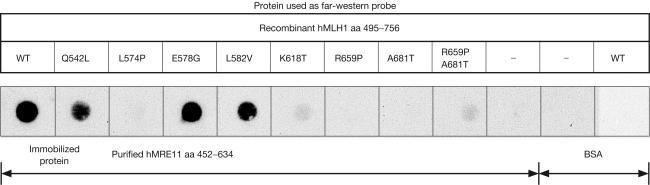
DNA mismatch repair (MMR) is essential in the surveillance of accurate transmission of genetic information, and defects in this pathway lead to microsatellite instability and hereditary nonpolyposis colorectal cancer (HNPCC). Our previous study raised the possibility that hMRE11 might be involved in MMR through physical interaction with hMLH1. Here, we show that hMRE11 deficiency leads to significant increase in MSI for both mono- and dinucleotide sequences. Furthermore, RNA-interference-mediated hMRE11-knockdown in HeLa cells results in MMR deficiency. Analysis of seven HNPCC-associated hMLH1 missense mutations located within the hMRE11-interacting domain shows that four mutations (L574P, K618T, R659P and A681T) cause near-complete disruption of the interaction between hMRE11 and hMLH1, and two mutations (Q542L and L582V) cause a 30% reduction of protein interaction. These findings indicate that hMRE11 represents a functional component of the MMR pathway and the disruption of hMLH1-hMRE11 interaction could be an alternative molecular explanation for hMLH1 mutations in a subset of HNPCC tumours.
Figures
Similar articles
-
The interplay between hMLH1 and hMRE11: role in MMR and the effect of hMLH1 mutations.Biochem Biophys Res Commun. 2008 May 30;370(2):338-43. doi: 10.1016/j.bbrc.2008.03.082. Epub 2008 Mar 26. Biochem Biophys Res Commun. 2008. PMID: 18373977 Free PMC article.
-
The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer.J Biol Chem. 1999 Mar 5;274(10):6336-41. doi: 10.1074/jbc.274.10.6336. J Biol Chem. 1999. PMID: 10037723
-
The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas.Am J Hum Genet. 2001 Oct;69(4):780-90. doi: 10.1086/323658. Epub 2001 Aug 24. Am J Hum Genet. 2001. PMID: 11524701 Free PMC article.
-
DNA mismatch repair defects: role in colorectal carcinogenesis.Biochimie. 2002 Jan;84(1):27-47. doi: 10.1016/s0300-9084(01)01362-1. Biochimie. 2002. PMID: 11900875 Review.
-
Causal link between microsatellite instability and hMRE11 dysfunction in human cancers.Mol Cancer Res. 2011 Nov;9(11):1443-8. doi: 10.1158/1541-7786.MCR-11-0322. Epub 2011 Aug 17. Mol Cancer Res. 2011. PMID: 21849470 Free PMC article. Review.
Cited by
-
The mismatch recognition protein MutSα promotes nascent strand degradation at stalled replication forks.Proc Natl Acad Sci U S A. 2022 Oct 4;119(40):e2201738119. doi: 10.1073/pnas.2201738119. Epub 2022 Sep 26. Proc Natl Acad Sci U S A. 2022. PMID: 36161943 Free PMC article.
-
Rad9 plays an important role in DNA mismatch repair through physical interaction with MLH1.Nucleic Acids Res. 2008 Nov;36(20):6406-17. doi: 10.1093/nar/gkn686. Epub 2008 Oct 8. Nucleic Acids Res. 2008. PMID: 18842633 Free PMC article.
-
Prognostic Significance of MRE11 Overexpression in Colorectal Cancer Patients.Cancers (Basel). 2023 Apr 24;15(9):2438. doi: 10.3390/cancers15092438. Cancers (Basel). 2023. PMID: 37173905 Free PMC article.
-
Eukaryotic Mismatch Repair in Relation to DNA Replication.Annu Rev Genet. 2015;49:291-313. doi: 10.1146/annurev-genet-112414-054722. Annu Rev Genet. 2015. PMID: 26436461 Free PMC article. Review.
-
Assessment of anti-recombination and double-strand break-induced gene conversion in human cells by a chromosomal reporter.J Biol Chem. 2012 Aug 24;287(35):29543-53. doi: 10.1074/jbc.M112.352302. Epub 2012 Jul 7. J Biol Chem. 2012. PMID: 22773873 Free PMC article.
References
-
- Carmell MA, Zhang L, Conklin DS, Hannon GJ, Rosenquist TA (2003) Germline transmission of RNAi in mice. Nat Struct Biol 10: 91–92 - PubMed
-
- Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P (2004) A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell 15: 31–41 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
