

Intricate targeting of immunoglobulin somatic hypermutation maximizes the efficiency of affinity maturation
- PMID: 15867095
- PMCID: PMC2213188
- DOI: 10.1084/jem.20042483
Intricate targeting of immunoglobulin somatic hypermutation maximizes the efficiency of affinity maturation
Abstract
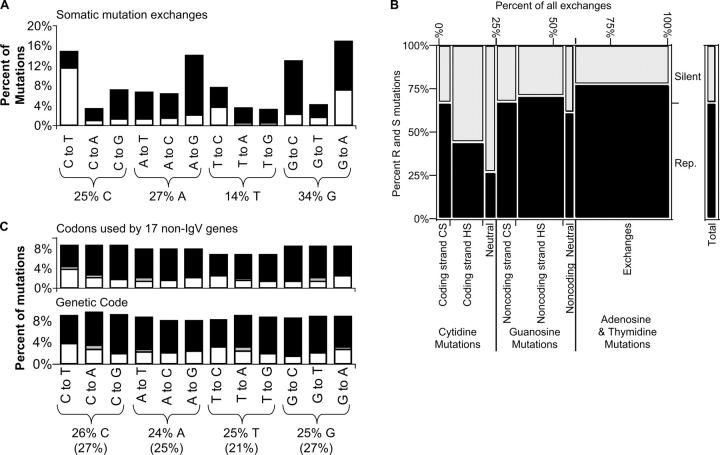
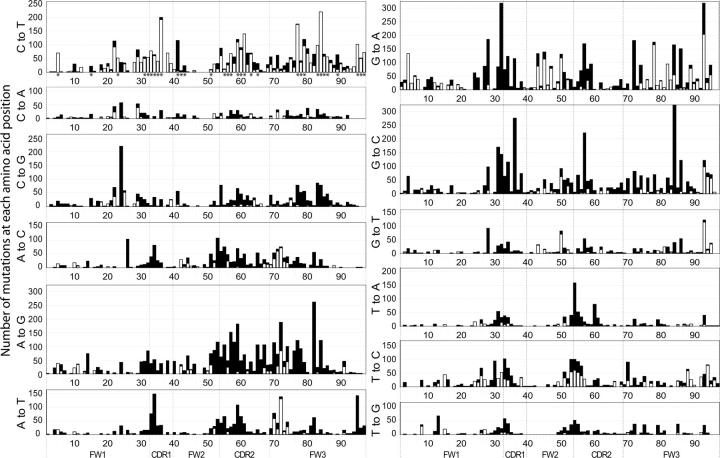
It is believed that immunoglobulin-variable region gene (IgV) somatic hypermutation (SHM) is initiated by activation-induced cytidine deaminase (AID) upon deamination of cytidine to deoxyuracil. Patch-excision repair of these lesions involving error prone DNA polymerases such as poleta causes mutations at all base positions. If not repaired, the deaminated nucleotides on the coding and noncoding strands result in C-to-T and G-to-A exchanges, respectively. Herein it is reported that IgV gene evolution has been considerably influenced by the need to accommodate extensive C deaminations and the resulting accumulation of C-to-T and G-to-A exchanges. Although seemingly counterintuitive, the precise placement of C and G nucleotides causes most C-to-T and G-to-A mutations to be silent or conservative. We hypothesize that without intricate positioning of C and G nucleotides the efficiency of affinity maturation would be significantly reduced due to a dominance of replacements caused by C and G transition mutations. The complexity of these evolved biases in codon use are compounded by the precise concomitant hotspot/coldspot targeting of AID activity and Poleta errors to maximize SHM in the CDRs and minimize mutations in the FWRs.
Figures


References
-
- Griffiths, G.M., C. Berek, M. Kaartinen, and C. Milstein. 1984. Somatic mutation and the maturation of immune response to 2-phenyl oxazolone. Nature. 312:271–275. - PubMed
-
- Rajewsky, K., I. Forster, and A. Cumano. 1987. Evolutionary and somatic selection of the antibody repertoire in the mouse. Science. 238:1088–1094. - PubMed
-
- Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. - PubMed
-
- Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell. 102:565–575. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
