

Calculation of absolute protein-ligand binding free energy from computer simulations
- PMID: 15867154
- PMCID: PMC1100764
- DOI: 10.1073/pnas.0409005102
Calculation of absolute protein-ligand binding free energy from computer simulations
Abstract
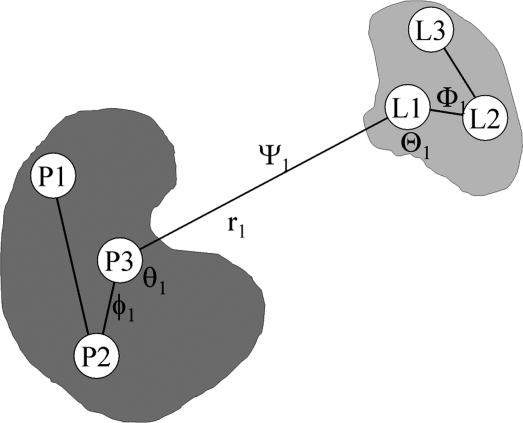
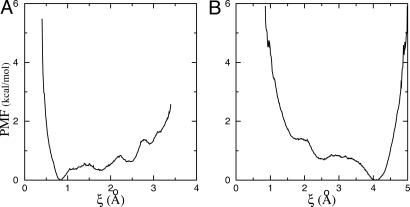
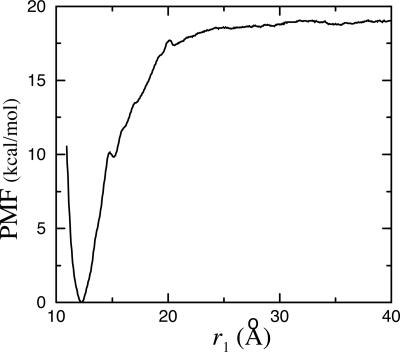
A general methodology for calculating the equilibrium binding constant of a flexible ligand to a protein receptor is formulated on the basis of potentials of mean force. The overall process is decomposed into several stages that can be computed separately: the free ligand in the bulk is first restrained into the conformation it adopts in the bound state, position, and orientation by applying biasing potentials, then it is translated into the binding site, where it is released completely. The conformational restraining potential is based on the root-mean-square deviation of the peptide coordinates relative to its average conformation in the bound complex. Free energy contributions from each stage are calculated by means of free energy perturbation potential of mean force techniques by using appropriate order parameters. The present approach avoids the need to decouple the ligand from its surrounding (bulk solvent and receptor protein) as is traditionally performed in the double-decoupling scheme. It is believed that the present formulation will be particularly useful when the solvation free energy of the ligand is very large. As an application, the equilibrium binding constant of the phosphotyrosine peptide pYEEI to the Src homology 2 domain of human Lck has been calculated. The results are in good agreement with experimental values.
Figures

References
-
- Wlodawer, A. (2002) Annu. Rev. Med. 53, 595–614. - PubMed
-
- Vindigni, A. (1999) Combinatorial Chem. High Throughput Scr. 2, 139–153. - PubMed
-
- Cheng, A. C., Calabro, V. & Frankel, A. D. (2001) Curr. Opin. Struct. Biol. 11, 478–484. - PubMed
-
- Garvie, C. W. & Wolberger, C. (2001) Mol. Cell. 8, 937–946. - PubMed
-
- Pawson, T. & Nash, P. (2003) Science 300, 445–452. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
