

PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints
- PMID: 15870257
- PMCID: PMC1132003
- DOI: 10.1101/gad.1291305
PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints
Abstract
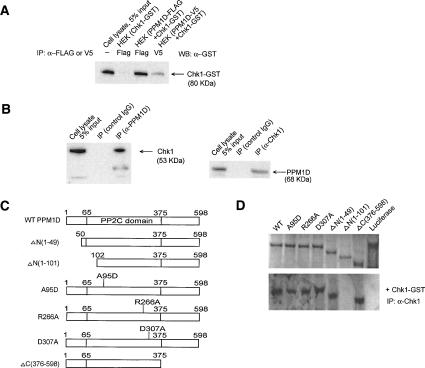
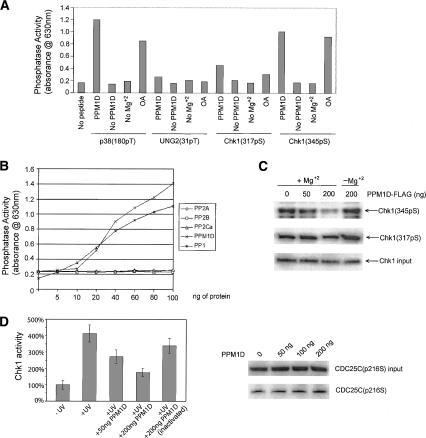
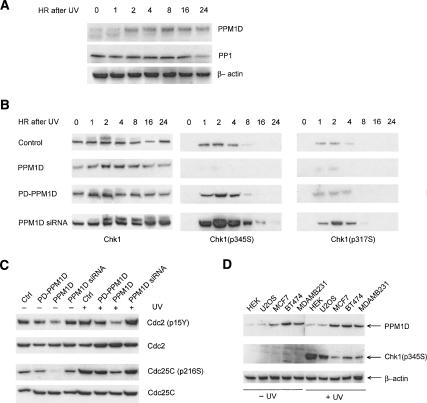
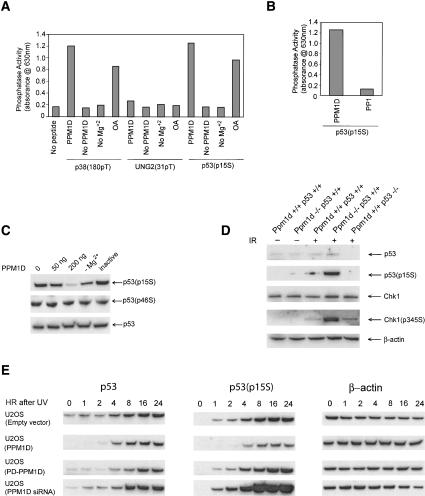
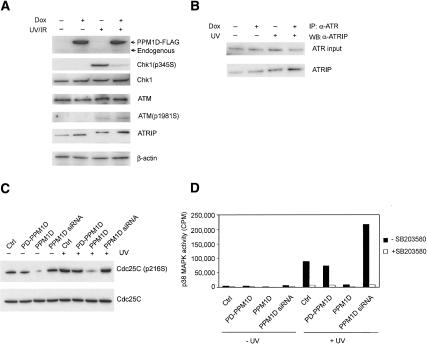
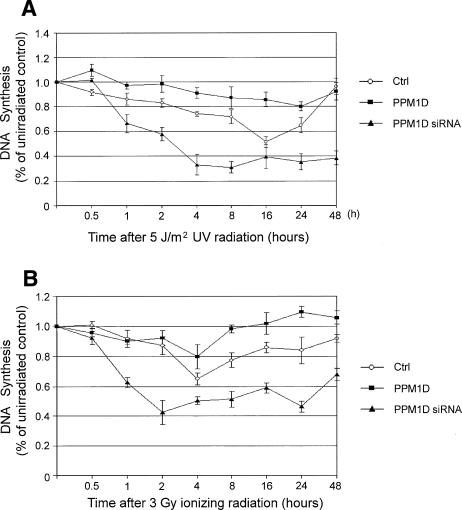
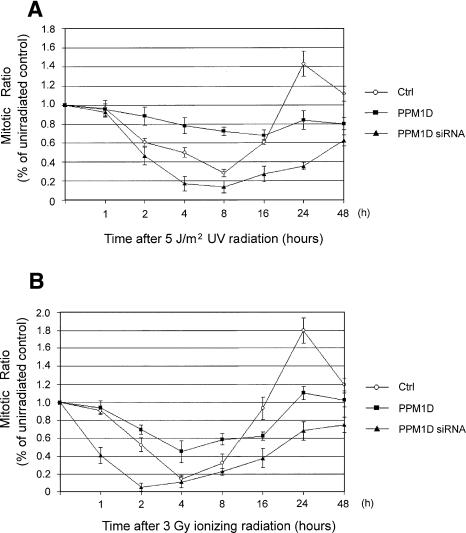
The ATM (ataxia-telangiectasia mutated) and ATR (ataxia-telangiectasia and Rad3-related) kinases respond to DNA damage by phosphorylating cellular target proteins that activate DNA repair pathways and cell cycle checkpoints in order to maintain genomic integrity. Here we show that the oncogenic p53-induced serine/threonine phosphatase, PPM1D (or Wip1), dephosphorylates two ATM/ATR targets, Chk1 and p53. PPM1D binds Chk1 and dephosphorylates the ATR-targeted phospho-Ser 345, leading to decreased Chk1 kinase activity. PPM1D also dephosphorylates p53 at phospho-Ser 15. PPM1D dephosphorylations are correlated with reduced cellular intra-S and G2/M checkpoint activity in response to DNA damage induced by ultraviolet and ionizing radiation. Thus, a primary function of PPM1D may be to reverse the p53 and Chk1-induced DNA damage and cell cycle checkpoint responses and return the cell to a homeostatic state following completion of DNA repair. These homeostatic functions may be partially responsible for the oncogenic effects of PPM1D when it is amplified and overexpressed in human tumors.
Figures
References
-
- Bakkenist C.J. and Kastan, M.B. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506. - PubMed
-
- ____. 2004a. Initiating cellular stress responses. Cell 118: 9–17. - PubMed
-
- ____. 2004b. Phosphatases join kinases in DNA-damage response pathways. Trends Cell Biol. 14: 339–341. - PubMed
-
- Banin S., Moyal, L., Shieh, S., Taya, Y., Anderson, C.W., Chessa, L., Smorodinsky, N.I., Prives, C., Reiss, Y., Shiloh, Y., et al. 1998. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous