

Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation
- PMID: 15870263
- PMCID: PMC1087725
- DOI: 10.1128/MCB.25.10.3886-3895.2005
Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation
Abstract
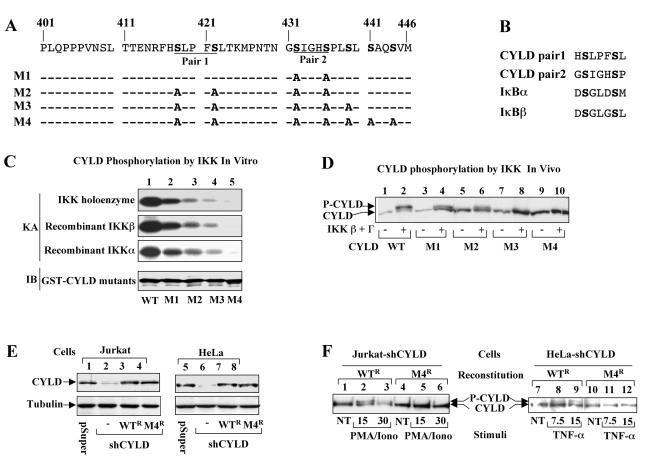
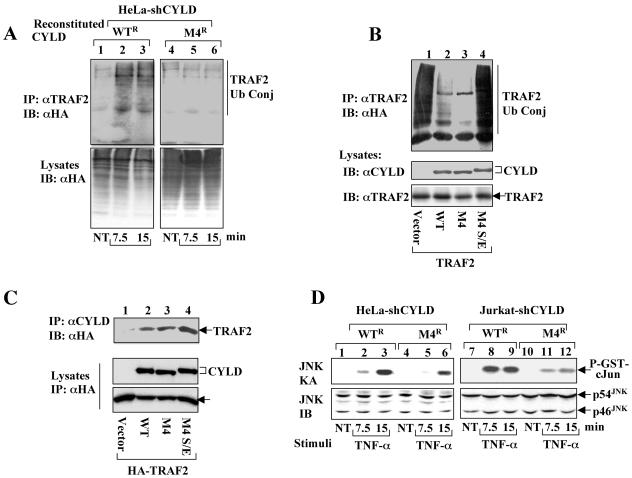
Tumor suppressor CYLD is a deubiquitinating enzyme (DUB) that inhibits the ubiquitination of key signaling molecules, including tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2). However, how the function of CYLD is regulated remains unknown. Here we provide evidence that inducible phosphorylation of CYLD is an important mechanism of its regulation. Under normal conditions, CYLD dominantly suppresses the ubiquitination of TRAF2. In response to cellular stimuli, CYLD undergoes rapid and transient phosphorylation, which is required for signal-induced TRAF2 ubiquitination and activation of downstream signaling events. Interestingly, the CYLD phosphorylation requires IkappaB kinase gamma (IKKgamma) and can be induced by IKK catalytic subunits. These findings suggest that CYLD serves as a novel target of IKK and that the site-specific phosphorylation of CYLD regulates its signaling function.
Figures





Similar articles
-
The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination.Nature. 2003 Aug 14;424(6950):801-5. doi: 10.1038/nature01802. Nature. 2003. PMID: 12917691
-
Negative regulation of JNK signaling by the tumor suppressor CYLD.J Biol Chem. 2004 Dec 31;279(53):55161-7. doi: 10.1074/jbc.M411049200. Epub 2004 Oct 20. J Biol Chem. 2004. PMID: 15496400
-
CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members.Nature. 2003 Aug 14;424(6950):793-6. doi: 10.1038/nature01803. Nature. 2003. PMID: 12917689
-
Regulation and function of IKK and IKK-related kinases.Sci STKE. 2006 Oct 17;2006(357):re13. doi: 10.1126/stke.3572006re13. Sci STKE. 2006. PMID: 17047224 Review.
-
Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity.Annu Rev Immunol. 2000;18:621-63. doi: 10.1146/annurev.immunol.18.1.621. Annu Rev Immunol. 2000. PMID: 10837071 Review.
Cited by
-
NEMO inhibits programmed necrosis in an NFκB-independent manner by restraining RIP1.PLoS One. 2012;7(7):e41238. doi: 10.1371/journal.pone.0041238. Epub 2012 Jul 26. PLoS One. 2012. PMID: 22848449 Free PMC article.
-
IκB kinase promotes Nrf2 ubiquitination and degradation by phosphorylating cylindromatosis, aggravating oxidative stress injury in obesity-related nephropathy.Mol Med. 2021 Oct 28;27(1):137. doi: 10.1186/s10020-021-00398-w. Mol Med. 2021. PMID: 34711178 Free PMC article.
-
SCFβ-TRCP regulates osteoclastogenesis via promoting CYLD ubiquitination.Oncotarget. 2014 Jun 30;5(12):4211-21. doi: 10.18632/oncotarget.1971. Oncotarget. 2014. PMID: 24961988 Free PMC article.
-
Inhibitory feedback control of NF-κB signalling in health and disease.Biochem J. 2021 Jul 16;478(13):2619-2664. doi: 10.1042/BCJ20210139. Biochem J. 2021. PMID: 34269817 Free PMC article. Review.
-
CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different.Cell Death Differ. 2017 Jul;24(7):1172-1183. doi: 10.1038/cdd.2017.46. Epub 2017 Mar 31. Cell Death Differ. 2017. PMID: 28362430 Free PMC article. Review.
References
-
- Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature 406:782-787. - PubMed
-
- Aggarwal, B. B. 2003. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3:745-756. - PubMed
-
- Anest, V., J. L. Hanson, P. C. Cogswell, K. A. Steinbrecher, B. D. Strahl, and A. S. Baldwin. 2003. Nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423:659-663. - PubMed
-
- Bignell, G. R., W. Warren, S. Seal, M. Takahashi, E. Rapley, R. Barfoot, H. Green, C. Brown, P. J. Biggs, S. R. Lakhani, C. Jones, J. Hansen, E. Blair, B. Hofmann, R. Siebert, G. Turner, D. G. Evans, C. Schrander-Stumpel, F. A. Beemer, A. van Den Ouweland, D. Halley, B. Delpech, M. G. Cleveland, I. Leigh, J. Leisti, and S. Rasmussen. 2000. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25:160-165. - PubMed
-
- Brooke, H. G. 1892. Epithelioma adenoides cysticum. Br. J. Dermatol. 4:269-287.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous