

Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency
- PMID: 15877279
- PMCID: PMC1196441
- DOI: 10.1086/430840
Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency
Abstract
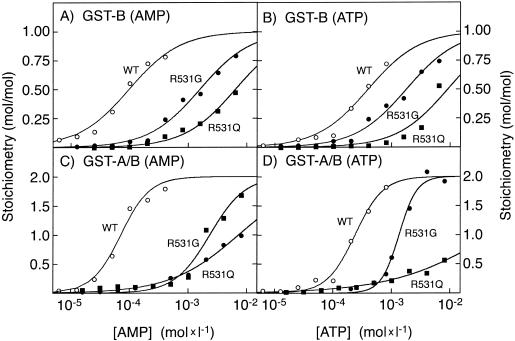
Fatal congenital nonlysosomal cardiac glycogenosis has been attributed to a subtype of phosphorylase kinase deficiency, but the underlying genes and mutations have not been identified. Analyzing four sporadic, unrelated patients, we found no mutations either in the eight genes encoding phosphorylase kinase subunits or in the two genes encoding the muscle and brain isoforms of glycogen phosphorylase. However, in three of five patients, we identified identical heterozygous R531Q missense mutations of the PRKAG2 gene, which encodes the gamma 2-subunit of AMP-activated protein kinase, a key regulator of energy balance. Biochemical characterization of the recombinant R531Q mutant protein showed >100-fold reduction of binding affinities for the regulatory nucleotides AMP and ATP but an enhanced basal activity and increased phosphorylation of the alpha -subunit. Other PRKAG2 missense mutations were previously identified in patients with autosomal dominant hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome, characterized by juvenile-to-adult clinical onset, moderate cardiac glycogenosis, disturbed excitation conduction, risk of sudden cardiac death in midlife, and molecular perturbations that are similar to--but less severe than--those observed for the R531Q mutation. Thus, recurrent heterozygous R531Q missense mutations in PRKAG2 give rise to a massive nonlysosomal cardiac glycogenosis of fetal symptomatic onset and rapidly fatal course, constituting a genotypically and clinically distinct variant of hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome. R531Q and other PRKAG2 mutations enhance the basal activity and alpha -subunit phosphorylation of AMP-activated protein kinase, explaining the dominant nature of PRKAG2 disease mutations. Since not all cases displayed PRKAG2 mutations, fatal congenital nonlysosomal cardiac glycogenosis seems to be genetically heterogeneous. However, the existence of a heart-specific primary phosphorylase kinase deficiency is questionable, because no phosphorylase kinase mutations were found.
Figures




Similar articles
-
Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase.Pediatr Res. 2007 Oct;62(4):499-504. doi: 10.1203/PDR.0b013e3181462b86. Pediatr Res. 2007. PMID: 17667862
-
Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy.J Clin Invest. 2002 Feb;109(3):357-62. doi: 10.1172/JCI14571. J Clin Invest. 2002. PMID: 11827995 Free PMC article.
-
A new mutation in PRKAG2 gene causing hypertrophic cardiomyopathy with conduction system disease and muscular glycogenosis.Neuromuscul Disord. 2006 Mar;16(3):178-82. doi: 10.1016/j.nmd.2005.12.004. Epub 2006 Feb 17. Neuromuscul Disord. 2006. PMID: 16487706
-
[AMP-activated protein kinase: how a mistake in energy gauge causes glycogen storage].Harefuah. 2007 Oct;146(10):770-5, 813-4. Harefuah. 2007. PMID: 17990392 Review. Hebrew.
-
Fatal infantile hypertrophic cardiomyopathy secondary to deficiency of heart specific phosphorylase b kinase.Virchows Arch A Pathol Anat Histopathol. 1993;423(4):303-7. doi: 10.1007/BF01606895. Virchows Arch A Pathol Anat Histopathol. 1993. PMID: 8236826 Review.
Cited by
-
A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway.Proc Natl Acad Sci U S A. 2006 Nov 14;103(46):17378-83. doi: 10.1073/pnas.0604708103. Epub 2006 Nov 3. Proc Natl Acad Sci U S A. 2006. PMID: 17085580 Free PMC article.
-
Glycogen metabolism and glycogen storage disorders.Ann Transl Med. 2018 Dec;6(24):474. doi: 10.21037/atm.2018.10.59. Ann Transl Med. 2018. PMID: 30740405 Free PMC article. Review.
-
PRKAG2 mutations presenting in infancy.J Inherit Metab Dis. 2017 Nov;40(6):823-830. doi: 10.1007/s10545-017-0072-0. Epub 2017 Aug 11. J Inherit Metab Dis. 2017. PMID: 28801758
-
Phenotypic expression and clinical outcomes in a South Asian PRKAG2 cardiomyopathy cohort.Sci Rep. 2020 Nov 26;10(1):20610. doi: 10.1038/s41598-020-77124-9. Sci Rep. 2020. PMID: 33244021 Free PMC article.
-
Shared genetic causes of cardiac hypertrophy in children and adults.N Engl J Med. 2008 May 1;358(18):1899-908. doi: 10.1056/NEJMoa075463. Epub 2008 Apr 9. N Engl J Med. 2008. PMID: 18403758 Free PMC article.
References
Electronic-Database Information
-
- American Type Culture Collection, http://www.atcc.org/
-
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for 1240C→G [accession number rs11231866], 907G→T [accession number rs3818199], and 1504G→A [accession number rs2227891])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Pompe disease, Danon disease, Cori or Forbes disease, Andersen disease, FHC/WPWS, and Phk deficiency)
References
-
- Antonarakis SE, Krawczak M, Cooper DN (2001) The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New York, pp 343–377
-
- Arad M, Maron BJ, Gorham JM, Johnson WH Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 352:362–372 - PubMed
-
- Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG (2003) Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107:2850–2856 - PubMed
-
- Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng, Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L (2004) The 5′-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem 279:38441–38447 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
